Notochord Remnant | Sacrum and Skull Base
- Notochordal origin - remnants of embryonic notochord
- Sacrum (50%), skull base (35%), mobile spine (15%)
- Brachyury = pathognomonic nuclear marker (T-box transcription factor)
- Wide resection is treatment of choice (intralesional = local recurrence)
- Slow-growing but locally aggressive with high recurrence
- “Physaliferous cells (bubbly cytoplasm) on histology
- “S100 and cytokeratin positive
- “Proton beam radiotherapy for unresectable
- “10-year survival approximately 40-60%
Notochord remnant tumor. The notochord normally regresses during development leaving only the nucleus pulposus. Chordoma arises from residual notochordal cells, explaining axial skeleton location.
Sacrum (50%): Most common. Skull base/clivus (35%): Second most common. Mobile spine (15%): Cervical most common mobile. Always midline/paracentral.
Physaliferous cells = pathognomonic (vacuolated, bubbly cytoplasm). Brachyury = nuclear marker specific for chordoma. Also S100+, cytokeratin+. Lobulated myxoid matrix.
Wide surgical resection is only chance for cure. Intralesional margins lead to local recurrence. Proton beam radiotherapy for unresectable or adjuvant. Chemotherapy limited role.
Overview and Epidemiology
Chordoma is a rare, slow-growing but locally aggressive malignant bone tumor arising from remnants of the embryonic notochord. It represents approximately 1-4% of all primary malignant bone tumors and accounts for approximately 20% of primary spine tumors.
Definition
Chordoma is a primary malignant bone tumor originating from notochordal remnants. Despite being classified as a low-grade malignancy based on histologic appearance, chordomas demonstrate locally aggressive behavior with high rates of local recurrence and potential for distant metastasis.
Epidemiology
Incidence: Rare tumor with annual incidence of approximately 0.08 per 100,000 population. Approximately 300 new cases diagnosed annually in the United States.
Age distribution: Typically presents in adults with peak incidence in the 5th to 7th decades (50-70 years). Can occur in children (approximately 5% of cases), where skull base location is more common.
Sex predilection: Male predominance with male-to-female ratio of approximately 2:1.
Location distribution:
- Sacrum: 50% (most common site)
- Skull base (clivus): 35% (second most common)
- Mobile spine: 15% (cervical greater than thoracic greater than lumbar)
The exclusive location in the axial skeleton reflects the embryologic distribution of notochord remnants.
Pathogenesis
The notochord is a transient embryonic structure that appears during the third week of gestation and serves as the primitive axial skeleton. It induces formation of the vertebral column and skull base, then normally undergoes complete regression by 8 weeks of gestation. The only normal remnant is the nucleus pulposus of intervertebral discs.
Chordoma arises from ectopic notochordal cell rests that persist after embryogenesis. These remnants are found in the axial skeleton, explaining why chordoma occurs exclusively in midline structures (sacrum, clivus, vertebrae). The tumor demonstrates notochordal differentiation with expression of brachyury, a T-box transcription factor essential for notochord development.
Genetics: Familial chordoma is rare but described. Duplications of the brachyury gene (TBXT) on chromosome 6q27 are associated with familial chordoma. Most cases are sporadic without identified hereditary pattern.
Pathophysiology
Molecular Biology
Brachyury expression: The defining molecular feature of chordoma is expression of brachyury (encoded by TBXT gene), a T-box transcription factor critical for notochord development. Brachyury is expressed in over 95% of chordomas and is considered pathognomonic. It is not expressed in morphologic mimics such as chondrosarcoma.
Genetic alterations: Chordomas demonstrate relatively simple karyotypes compared to other sarcomas. Common alterations include:
- Loss of chromosome 1p (associated with worse prognosis)
- Loss of 9p (CDKN2A/p16 locus)
- Amplification or mutation of EGFR and PDGFR pathways
- TP53 mutations in some cases
Growth characteristics: Despite low mitotic rate and bland cytologic features, chordomas are locally aggressive. They demonstrate:
- Slow but persistent growth
- Local invasion of bone and soft tissue
- Entrapment of neurovascular structures
- High local recurrence rates (30-50% even with adequate resection)
- Late distant metastasis potential (10-40%, usually to lungs, bone, liver)
Histological Subtypes
Conventional (classic) chordoma: 80% of cases. Lobulated architecture with cords and sheets of tumor cells in abundant myxoid matrix. Characteristic physaliferous cells (physaliphorous = bubble-bearing) are large cells with vacuolated, bubbly cytoplasm containing glycogen and mucin. Nuclei are typically small, round, and bland. Mitotic figures are rare.
Chondroid chordoma: 5-15% of cases. Contains areas of hyaline cartilage in addition to typical chordoma features. More common in skull base. Historically considered to have better prognosis, though recent studies suggest similar outcomes to conventional type when adequately treated. Must distinguish from chondrosarcoma using brachyury immunostaining.
Dedifferentiated chordoma: Less than 5% of cases. Contains areas of high-grade sarcomatous transformation with increased cellularity, atypia, and mitotic activity. Worst prognosis with more aggressive behavior and higher metastatic potential. Can occur de novo or in recurrent tumors. Median survival approximately 1 year.
Poorly differentiated chordoma: Rare variant seen primarily in children and young adults. Lacks typical physaliferous cells. More aggressive clinical course.
Histopathology and Diagnosis
Gross Pathology
Lobulated, gelatinous gray-white to tan mass. Cut surface demonstrates mucoid, glistening appearance. Hemorrhage and necrosis uncommon except in dedifferentiated variants. Soft tissue component often extensive.
Microscopic Features

Architecture: Lobular growth pattern separated by fibrous septae. Cords, nests, and sheets of tumor cells in abundant extracellular myxoid matrix.
Cytology:
- Physaliferous cells: Pathognomonic feature. Large cells with abundant, vacuolated, bubbly cytoplasm (vacuoles contain mucin and glycogen). Cytoplasmic vacuoles push nucleus to periphery creating signet-ring appearance in some cells.
- Nuclei: Small, round to oval, centrally or eccentrically placed. Minimal pleomorphism in conventional type.
- Mitotic activity: Rare mitotic figures (fewer than 1 per 10 high-power fields in conventional type).
Matrix: Abundant extracellular myxoid/mucoid matrix rich in proteoglycans. Matrix stains with Alcian blue (blue) and mucicarmine (pink).
Immunohistochemistry
Brachyury (T-brachyury): DIAGNOSTIC MARKER. Nuclear expression in over 95% of chordomas. Highly specific - positive only in chordoma and normal notochord. Negative in chondrosarcoma and other mimics. This is the single most important immunostain to confirm diagnosis.
S100 protein: Positive in 80-100% of cases. Strong, diffuse nuclear and cytoplasmic staining. Also positive in chondrosarcoma (thus not specific).
Cytokeratin: Positive in 80-100%. Includes low molecular weight keratins (CAM 5.2, AE1/AE3). Dot-like paranuclear pattern characteristic.
EMA (epithelial membrane antigen): Positive in most cases (70-90%).
Vimentin: Variably positive.
NEGATIVE stains: Desmin, smooth muscle actin, chromogranin, synaptophysin (helps exclude other tumor types).
Differential Diagnosis
- Key feature
- Physaliferous cells, lobular myxoid matrix, midline
- Brachyury
- Positive (nuclear)
- Cytokeratin
- Positive (dot-like)
- Other markers
- S100+, EMA+
- Key feature
- Malignant chondrocytes in hyaline cartilage
- Brachyury
- Negative
- Cytokeratin
- Negative
- Other markers
- S100+ (not discriminatory)
- Key feature
- Glandular/epithelial pattern, known primary
- Brachyury
- Negative
- Cytokeratin
- Positive
- Other markers
- Site-specific markers (TTF1, CDX2 etc)
- Key feature
- Sacrococcygeal, perivascular myxoid pseudorosettes
- Brachyury
- Negative
- Cytokeratin
- Negative
- Other markers
- GFAP+
- Key feature
- Benign clival notochordal rest, under 2 cm, no bone destruction
- Brachyury
- Positive
- Cytokeratin
- Positive
- Other markers
- Identical histology - distinguished by size/imaging
Chondrosarcoma: Most important differential, especially for chondroid chordoma. Both can have cartilaginous matrix and S100 positivity. Brachyury distinguishes: positive in chordoma, negative in chondrosarcoma. Cytokeratin also typically negative in chondrosarcoma.
Metastatic carcinoma: Axial skeleton location and cytokeratin positivity can mimic metastatic adenocarcinoma. Brachyury negative in carcinomas. Clinical history and additional immunostains help.
Myxopapillary ependymoma: Occurs in sacrococcygeal region, can have myxoid matrix. Lacks physaliferous cells and brachyury positivity. GFAP positive (negative in chordoma).
Ecchordosis physaliphora: Benign notochordal remnant typically found incidentally at clivus. Small (less than 2 cm), no bone destruction, identical histology to chordoma. Clinical and imaging features distinguish.
Clinical Presentation
Sacral Chordoma
Presents with nonspecific symptoms leading to delayed diagnosis. Average symptom duration before diagnosis is 6-12 months.
Pain: Most common presenting symptom (70-80% of patients). Initially mild, intermittent sacral or coccygeal pain. Progressively worsens and becomes constant. May radiate to buttocks, hips, or lower extremities.
Neurological symptoms: Occur in approximately 50% at presentation. Include:
- Radicular pain (L5, S1 dermatomes most common)
- Motor weakness (foot drop from L5 involvement)
- Sensory changes in saddle distribution
- Bowel dysfunction (constipation most common)
- Bladder dysfunction (urinary retention or incontinence)
- Sexual dysfunction
Pelvic mass: Large tumors may cause palpable presacral mass on rectal examination. Anterior extension can cause:
- Constipation or change in bowel habits
- Urinary symptoms from bladder compression
- Dyspareunia in women
- Rectal bleeding (rare, suggests mucosal invasion)
Skull Base Chordoma
Presents earlier due to critical location.
Cranial nerve palsies: Most common presentation (60-70%). Abducens nerve (CN VI) most commonly affected, causing diplopia. Other cranial nerves can be involved depending on extension.
Headache: Present in 40-50% of patients. Typically retro-orbital or vertex headache.
Visual symptoms: Diplopia, visual field defects, decreased visual acuity.
Endocrine dysfunction: Large tumors can affect pituitary function causing hypopituitarism.
Nasal/pharyngeal symptoms: Nasal obstruction, epistaxis, dysphagia with anterior extension.
Mobile Spine Chordoma
Neck or back pain: Localized pain at tumor site. Initially mechanical, progressively worsens.
Radiculopathy: Nerve root compression causes dermatomal pain and neurological deficits.
Myelopathy: Spinal cord compression causes:
- Upper motor neuron signs below level of lesion
- Gait disturbance
- Bladder and bowel dysfunction
- Sensory level
Physical Examination
Inspection: May reveal visible/palpable mass in advanced cases.
Neurological examination:
- Comprehensive motor examination (look for lower motor neuron signs from nerve root involvement)
- Sensory testing (dermatomes, saddle anesthesia)
- Reflexes (may be diminished with nerve root compression)
- Rectal tone assessment
- Perianal sensation
Rectal examination: Essential for sacral lesions. May palpate presacral mass - typically firm, fixed, smooth or lobulated.
Investigations
Laboratory Studies
Routine labs: Generally normal. CBC, basic metabolic panel to assess baseline function.
Tumor markers: No specific serum markers for chordoma. Alkaline phosphatase may be elevated but is nonspecific.
Imaging
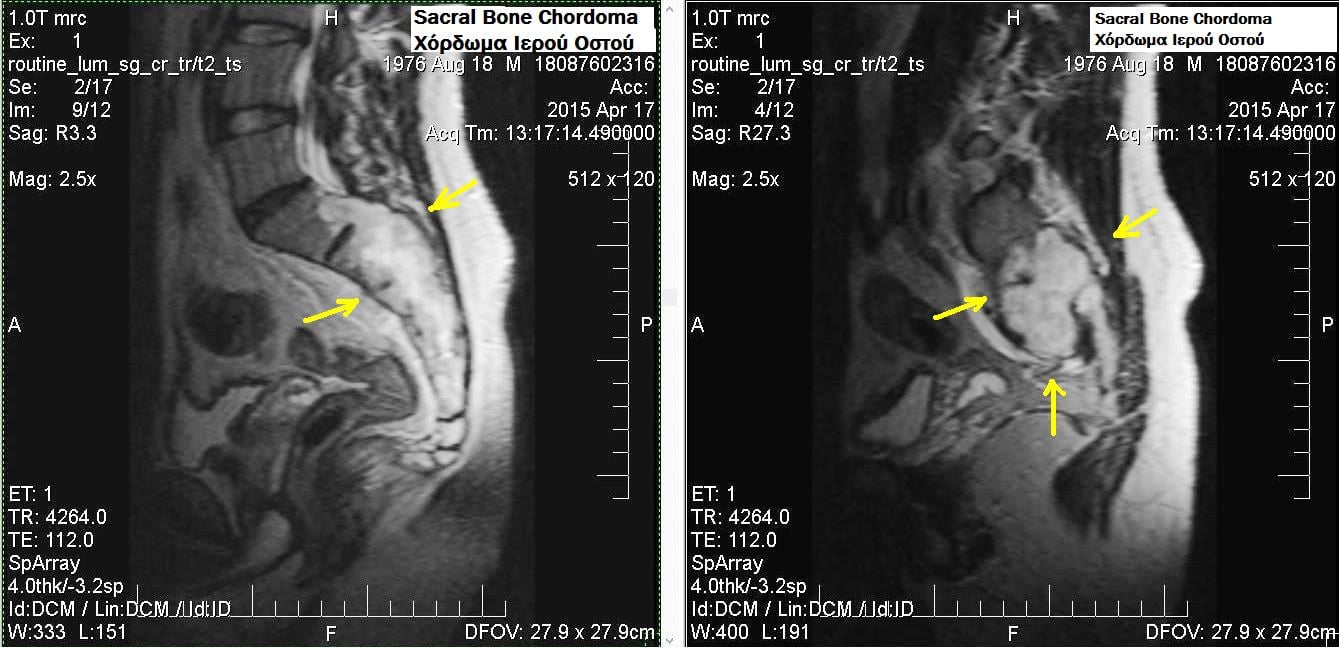
MRI is the gold standard for chordoma assessment. Provides superior soft tissue detail, neural structure visualization, and extent of marrow involvement.
Typical MRI characteristics:
- T1-weighted: Hypointense to isointense relative to muscle (due to high water content in myxoid matrix)
- T2-weighted: Very hyperintense (bright signal from abundant mucoid/myxoid matrix)
- T1 post-contrast: Heterogeneous enhancement with irregular pattern
- Location: Midline or paracentral destructive lesion
- Margins: Lobulated contour with internal septations
- Soft tissue component: Usually large, often exceeds bony component
Sequences to obtain:
- Sagittal T1 and T2 of entire spine (assess skip lesions)
- Axial T1 and T2 through lesion
- Coronal STIR or fat-suppressed T2
- T1 post-gadolinium with fat saturation
MRI is essential for:
- Defining extent of tumor
- Assessing involvement of adjacent structures (dura, nerve roots, vessels)
- Surgical planning (relationship to critical structures)
- Post-treatment surveillance for recurrence
MRI provides the most comprehensive assessment for chordoma diagnosis and management planning.
Tissue Diagnosis
Biopsy indications: Tissue diagnosis mandatory before definitive treatment. Clinical and imaging features alone cannot definitively diagnose chordoma.
Biopsy technique: Image-guided core needle biopsy preferred (CT or fluoroscopy guidance). Considerations:
- Plan biopsy tract to allow en bloc excision with definitive surgery
- Avoid contaminating uninvolved tissue planes
- Obtain adequate tissue for histology and immunohistochemistry
- Posterior approach typically used for sacral lesions
- Send fresh tissue if molecular studies planned
Open biopsy: Reserved for cases where core biopsy nondiagnostic or inadequate. Risks tumor contamination of surgical field.
Staging Investigations
Chest imaging: Chest CT required to exclude pulmonary metastases.
Whole spine imaging: MRI of entire spine to assess for skip metastases (rare but described).
Skeletal survey or bone scan: Consider if symptoms suggest other skeletal involvement.
Laboratory: Baseline renal and hepatic function for treatment planning.
Management Algorithm
Core Principles
Wide surgical resection is the only curative treatment. The margin achieved is the single most important prognostic factor for local control and survival. Intralesional surgery (tumor violation during resection) results in 50-90% local recurrence.
Goals of surgery:
- En bloc resection with wide margins (negative margins on all surfaces)
- Preservation of neurologic function when possible
- Maintain structural stability
- Minimize morbidity
Radiation therapy role: Used when wide resection not feasible or for positive margins. Proton beam or carbon ion therapy preferred over conventional photon radiation.
Chemotherapy: No proven role for conventional chemotherapy. Chordomas are not chemosensitive.
These principles apply to all chordoma treatment decisions.
Radiation Therapy
Conventional Photon Radiation
Chordomas are relatively radioresistant to conventional photon radiation. Doses required for control (greater than 70 Gy) exceed tolerance of surrounding normal tissues (brainstem, spinal cord, bowel, bladder). Therefore, conventional radiation has limited efficacy as primary treatment but may be used in palliative settings.
Proton Beam Therapy
Mechanism: Protons deposit maximum energy at a specific depth (Bragg peak) with minimal exit dose. This allows delivery of high radiation doses to tumor while sparing adjacent normal tissues.
Indications:
- Unresectable chordomas (skull base, sacral with major vascular involvement)
- Positive margins after surgery (adjuvant)
- Recurrent tumors not amenable to further surgery
- Patient choice when surgery declined
Dosing: Typical doses 70-80 Gy (RBE) in 35-40 fractions.
Outcomes:
- 5-year local control: 60-80% for skull base chordomas
- 5-year local control: 50-70% for sacral chordomas
- Best results when combined with maximal safe surgery
Availability: Limited to specialized centers with proton beam facilities.
Carbon Ion Therapy
Similar concept to proton therapy but with theoretical radiobiologic advantages. Available at even fewer centers worldwide. Emerging evidence suggests similar or slightly better local control compared to proton therapy.
Conventional Radiation Role
Palliative treatment: Pain control, symptom management in metastatic disease or patients not candidates for surgery/proton therapy.
Adjuvant after intralesional surgery: May reduce recurrence risk but inferior to proton therapy.
Systemic Therapy
Chemotherapy
Conventional cytotoxic chemotherapy has no established role in chordoma management. Chordomas are not chemosensitive. Multiple agents tested in clinical trials (ifosfamide, doxorubicin, cisplatin, temozolomide) with minimal response rates.
Targeted Therapies
Based on molecular profiling showing activation of receptor tyrosine kinase pathways, several targeted agents have been investigated:
Imatinib: PDGFR inhibitor. Small series showed disease stabilization in some patients but no objective responses. May have role in metastatic/unresectable disease.
EGFR inhibitors (erlotinib, cetuximab): Occasional responses reported in case reports. No large trials.
mTOR inhibitors: Under investigation based on pathway activation in some chordomas.
Immunotherapy: Checkpoint inhibitors being explored. Response rates appear low given low mutational burden of chordomas. Brachyury-targeted vaccine in clinical trials.
Current Approach to Systemic Therapy
No standard systemic therapy exists. For patients with metastatic or unresectable progressive disease, consider:
- Clinical trial enrollment (preferred)
- Targeted therapy (imatinib or EGFR inhibitor) with understanding of limited evidence
- Palliative care focus
Surgical Management
Sacral Chordoma Surgery
Preoperative planning:
- Define tumor extent with MRI (relationship to S2-S3 junction critical)
- Assess vascular anatomy (iliac vessels, presacral plexus)
- Counsel patient on functional outcomes (bowel, bladder, sexual function)
- Optimize medical status
- Consider preoperative embolization for large hypervascular tumors
Functional considerations:
- S1 nerve roots: Bilateral preservation essential for walking. Unilateral sacrifice causes foot drop.
- S2 nerve roots: Bilateral preservation maintains bladder and bowel function. Bilateral sacrifice requires permanent catheterization and often colostomy.
- S3 and below: Sacrifice causes varying degrees of dysfunction but generally better tolerated than S2.
High sacrectomy (above S2-S3): Requires sacrifice of S2-S3 roots with significant functional impact. Reserved for high tumors where necessary for margins.
Low sacrectomy (S3 and below): Preserves S1-S2 roots with better functional outcomes. May still have bowel/bladder/sexual dysfunction but less severe.
Surgical approach:
Combined anterior-posterior approach: Most common for large sacral chordomas.
- Anterior phase: Abdominal/pelvic approach. Mobilize rectum, ligate vessels, dissect presacral component.
- Posterior phase: Patient repositioned prone. Complete osteotomy and remove specimen en bloc.
- Staged procedures (anterior first, posterior days later) or single-stage both described.
Posterior-only approach: Feasible for smaller tumors without large anterior extension.
Technique pearls:
- Use frozen sections to confirm negative margins
- Preserve as many nerve roots as feasible while achieving negative margins
- Lumbopelvic reconstruction needed for high sacrectomy (spinopelvic fixation)
- Soft tissue coverage critical (may need flap reconstruction)
- Plan biopsy tract excision en bloc with specimen
Outcomes:
- 5-year local recurrence-free survival: 60-80% with wide margins
- 5-year local recurrence-free survival: 20-40% with intralesional margins
- Overall survival: 60-80% at 5 years, 40-60% at 10 years
There is good evidence that margin status predicts outcomes, with adequate surgical resection being the cornerstone of treatment.
Complications
Surgical Complications
Intraoperative:
- Hemorrhage: Sacral resections can involve significant blood loss (presacral venous plexus, iliac vessels). Blood transfusion commonly required.
- Dural tear: High risk in spine cases. CSF leak risk.
- Vascular injury: Iliac vessels at risk in sacral surgery. Requires vascular surgery availability.
- Nerve injury: Intentional sacrifice versus inadvertent injury to roots intended to preserve.
Early postoperative:
- Wound complications: Infection, dehiscence, seroma. High rate (20-40%) given large dead space and possible contamination.
- Deep infection: Potential for infection of hardware/reconstruction. May require removal and prolonged antibiotics.
- Neurologic deficits: Lower extremity weakness, bladder/bowel dysfunction if nerve roots sacrificed or injured.
- Venous thromboembolism: High risk due to pelvic surgery and immobility.
Late complications:
- Chronic pain: Neuropathic pain common, especially with nerve root sacrifice.
- Bladder dysfunction: Requires intermittent catheterization or indwelling catheter if bilateral S2 sacrifice.
- Bowel dysfunction: Constipation most common. May require bowel regimen or colostomy if severe.
- Sexual dysfunction: Erectile dysfunction in men, dyspareunia in women.
- Implant failure: Spinopelvic fixation failure can occur (5-10% of cases).
- Sacral insufficiency: Stress fractures of remaining sacrum.
Radiation Complications
Acute: Skin erythema, mucositis (skull base), diarrhea (sacral).
Late:
- Radiation necrosis: Brain/brainstem necrosis (skull base tumors), bowel injury (sacral tumors)
- Secondary malignancy: Long-term risk of radiation-induced sarcoma
- Endocrine dysfunction: Hypopituitarism after skull base radiation
- Neurologic deficits: Cranial neuropathies, myelopathy risk
Proton/carbon ion therapy reduces risk of late complications compared to conventional radiation due to normal tissue sparing.
Disease-Related Complications
Local progression: Pain, neurologic deterioration, mass effect on pelvic organs.
Metastatic disease: Occurs in 10-40% of patients, usually late in disease course. Lungs most common site, followed by bone and liver. Median time to metastasis is 5-7 years after diagnosis.
Prognosis and Follow-Up
Prognostic Factors
Margin status: Single most important factor. Wide negative margins associated with significantly better local control and survival.
- Wide margins: 5-year local recurrence 10-30%
- Marginal/intralesional: 5-year local recurrence 50-90%
Tumor location:
- Sacral: Generally better prognosis (more amenable to wide resection)
- Skull base: More challenging to achieve margins, often require adjuvant radiation
- Mobile spine: Intermediate prognosis
Histologic subtype:
- Conventional: Standard prognosis
- Chondroid: Similar to conventional when adequately treated
- Dedifferentiated: Worst prognosis (median survival approximately 1 year)
Tumor size: Larger tumors associated with worse outcomes (harder to achieve margins, more likely to involve critical structures).
Age: Younger age generally associated with better survival.
Recurrence status: Recurrent tumors have worse prognosis than primary tumors.
Survival Data
5-year overall survival: 65-80% 10-year overall survival: 40-60% 5-year progression-free survival: 50-70% with optimal treatment
Cause of death: Most patients who die of chordoma do so from local progression rather than distant metastases. Uncontrolled local disease in sacrum or skull base causes significant morbidity.
Surveillance Protocol
Imaging schedule:
- Years 1-2: MRI of primary site every 3-4 months
- Years 3-5: MRI every 6 months
- After 5 years: Annual MRI
- Chest CT: Annually to monitor for pulmonary metastases
- Whole body imaging: Consider if symptoms suggest new disease sites
Clinical follow-up: Assess for symptoms of local recurrence, neurologic function, pain control, functional status (bowel/bladder).
Long-term surveillance: Lifelong follow-up recommended as late recurrences occur (can recur 10-20 years after initial treatment).
Poorly Differentiated (SMARCB1/INI1-Deficient) Chordoma
The Histological Subtypes section names a "poorly differentiated chordoma... seen primarily in children and young adults... lacks typical physaliferous cells... more aggressive", but never gives its modern molecular definition - a distinct, aggressive entity now defined by loss of SMARCB1 (INI1).
- The defining lesion. Poorly differentiated chordoma is characterised by loss of nuclear SMARCB1 (INI1) expression on immunohistochemistry (deletion/inactivation of the SMARCB1 gene on 22q11) - the same tumour-suppressor lost in atypical teratoid/rhabdoid tumour and epithelioid sarcoma. It remains brachyury-positive (confirming notochordal origin) but shows solid, epithelioid/rhabdoid sheets without the classic physaliphorous cells or myxoid matrix.
- Who and where. It occurs disproportionately in children and young adults and favours the skull base and cervical spine (clival/cervical), unlike the sacral predominance of conventional chordoma.
- Why it matters. It is substantially more aggressive than conventional chordoma, with earlier metastasis and shorter survival, so recognising INI1 loss changes prognosis and surveillance and flags a paediatric skull-base tumour that behaves like a high-grade malignancy. Its shared SMARCB1 biology has made it a candidate for EZH2 inhibition (as trialled in other SMARCB1-deficient tumours), one of the few rational targeted avenues in chordoma.
- The pitfall. Because it lacks physaliphorous cells and myxoid matrix it is easily mistaken for another poorly differentiated round-cell or rhabdoid tumour - brachyury positivity plus INI1 loss is the combination that secures the diagnosis.
Q: A child has an aggressive clival/cervical tumour lacking physaliphorous cells - how is poorly differentiated chordoma confirmed and why does it matter? A: It is brachyury-positive (notochordal) but shows loss of nuclear SMARCB1/INI1 on IHC (22q11) - the combination that secures the diagnosis despite the absent physaliphorous cells and myxoid matrix. It is a paediatric skull-base/cervical entity that is far more aggressive (earlier metastasis, shorter survival) than conventional chordoma, and its SMARCB1 loss makes it a candidate for EZH2 inhibition.
Sacrectomy Functional Outcomes by Nerve-Root Level (Gunterberg Rule)
The topic and its Fourney evidence card repeatedly turn on which sacral nerve roots are preserved, but never give the crisp classic rule (Gunterberg; Todd) that predicts continence and ambulation from the level of the osteotomy - the single most important thing to counsel a patient before sacrectomy.
- Ambulation (S1). The S1 roots carry the main motor supply to the gastrocnemius-soleus and gluteals; bilateral S1 sacrifice causes major weakness (foot drop, loss of push-off and hip extension), so preserving at least the S1 roots is the threshold for useful walking.
- Continence (S2-S3, the key rule). Bladder, bowel and sexual function depend on the S2-S3-S4 parasympathetic outflow. Classic teaching: bilateral preservation of S1-S2-S3 gives near-normal function; preserving BOTH S3 roots (resecting below S3) usually preserves continence; retaining even ONE S3 root often maintains acceptable bladder/bowel control; but bilateral sacrifice of S2 and above reliably produces a neurogenic bladder needing catheterisation and bowel dysfunction (often a colostomy).
- The unilateral rule. Because these roots are partly redundant across the midline, unilateral sacrifice of the sacral roots (a hemisacrectomy sparing the contralateral roots) generally preserves bladder, bowel and sexual function - it is bilateral sacrifice at a given level that is functionally decisive.
- Counselling. This is why the S2-S3 junction is the pivotal planning line on MRI: a resection that must cross above both S3 roots commits the patient to bladder, bowel and sexual dysfunction, and weighing that against margin adequacy is the central pre-operative conversation (margins should not be compromised for function when they are needed for cure).
Q: Which sacral roots must be preserved for continence and for walking after sacrectomy? A: Walking needs the S1 roots (bilateral S1 loss → foot drop and gluteal weakness). Continence needs the S2-S3 parasympathetic outflow: preserving both S3 roots usually keeps continence, and even one retained S3 root often maintains acceptable control, whereas bilateral sacrifice of S2 and above gives a neurogenic bladder (catheterisation) and bowel dysfunction (often colostomy). Unilateral sacral sacrifice generally spares function - it is bilateral loss that is decisive, which is why the S2-S3 junction is the key planning line.
Guidelines, Registries & Global Practice
Global Epidemiology
Chordoma is uniformly rare worldwide, with a population-based incidence of approximately 0.08 per 100,000 (SEER, McMaster 2001). There is a consistent male predominance (~1.7:1) and a peak in the fifth to seventh decades, with cranial disease over-represented in younger patients and children. Population (SEER) data show a roughly even axial distribution (cranial ~32%, mobile spine ~33%, sacrum ~29%), whereas surgical series quote the classic sacrum 50% / skull base 35% / mobile spine 15% because surgically resected sacral tumours are over-represented.
Side-by-Side Guideline Comparison
- Surgery
- En bloc resection with wide margins at a reference centre
- Radiation
- High-dose particle therapy (proton/carbon ion) if margins inadequate or unresectable
- Systemic / Other
- No standard chemo; favour clinical trials and molecular profiling
- Surgery
- Wide en bloc resection is the goal; avoid intralesional violation
- Radiation
- Definitive high-dose RT when surgery would be mutilating or incomplete
- Systemic / Other
- Imatinib/EGFR agents only within trial or compassionate use
- Surgery
- Wide excision; spondylectomy/sacrectomy by expert teams
- Radiation
- RT (proton preferred) for residual, recurrent or unresectable disease
- Systemic / Other
- Targeted therapy or trial for progressive systemic disease
- Surgery
- Centralised management in designated sarcoma/spinal tumour MDTs
- Radiation
- Access to proton therapy via national high-energy proton service
- Systemic / Other
- Trial enrolment; no routine cytotoxic chemotherapy
The unifying message across all bodies is identical: wide en bloc resection at a high-volume reference centre, with high-dose particle radiotherapy reserved for inadequate margins or unresectable disease, and no role for routine cytotoxic chemotherapy.
Registry and Reference-Centre Notes
- Chordoma is too rare for arthroplasty-style implant registries; the most robust outcome data come from national sarcoma databases and reference-centre series (e.g. Rizzoli, MD Anderson, Massachusetts General, Swedish Musculoskeletal Tumour Centre).
- The Chordoma Foundation maintains an international patient registry and biobank that increasingly drives multi-centre collaborative evidence.
High- vs Limited-Resource Practice Variation
- Particle therapy (proton/carbon ion) is concentrated in a small number of centres in North America, Europe and East Asia; many regions require interstate or international referral, creating access inequity and treatment delay.
- In limited-resource settings, conventional photon radiotherapy and the quality of the index surgical resection carry disproportionate weight, since a contaminated first operation cannot be undone and dominates lifelong local-control prospects.
- Early referral before biopsy to a specialist tumour centre is the single most transferable, resource-independent quality measure, ensuring a correctly planned, excisable biopsy tract.
Controversies & Areas of Uncertainty
-
Proton versus carbon ion therapy: Both spare normal tissue via the Bragg peak, but no randomised trial compares them. Carbon ions carry a higher relative biological effectiveness and may suit radioresistant tumours, yet are available at very few centres. Choice is largely driven by access rather than proven superiority.
-
Definitive radiotherapy versus mutilating surgery: For high sacral or skull-base tumours where wide resection would cost bladder, bowel and ambulatory function, the threshold for offering definitive high-dose particle therapy instead of en bloc resection remains a genuine MDT judgement, not a settled algorithm.
-
Prognostic weight of the chondroid variant: Historically considered favourable, but several modern series suggest outcomes equal to conventional chordoma once margin and dose are controlled for. The variant is now seen more as a histological than a prognostic distinction.
-
Role of systemic therapy: Imatinib gives disease stabilisation (clinical benefit ~64%) but almost no objective shrinkage (ORR 2% in the phase II trial). EGFR inhibitors, brachyury-targeted vaccines and immune checkpoint blockade remain investigational; none is standard of care.
-
Optimal surveillance duration: Late recurrences and metastases (well beyond 10 years) mean the ideal length and intensity of lifelong imaging follow-up is not evidence-defined and varies between centres.
MCQ Practice Points
Q: What is the embryologic origin of chordoma?
A: Notochordal remnants. The notochord is an embryonic structure that forms the primitive axial skeleton and normally regresses completely by 8 weeks of gestation, leaving only the nucleus pulposus. Chordoma arises from ectopic notochordal cell rests that persist after embryogenesis. This explains the exclusive location of chordoma in midline axial skeleton structures.
Q: What is the pathognomonic immunohistochemical marker for chordoma?
A: Brachyury - a nuclear T-box transcription factor encoded by the TBXT gene. Brachyury is positive in over 95% of chordomas and is the single most specific marker. It is negative in chondrosarcoma and other differential diagnoses. Also positive: S100 (80-100%), cytokeratin (80-100%). The combination of brachyury, S100, and cytokeratin positivity confirms chordoma.
Q: What is the characteristic cell type in chordoma histology?
A: Physaliferous cells - large cells with abundant vacuolated, bubbly cytoplasm containing glycogen and mucin. The cytoplasmic vacuoles push the nucleus to the periphery, sometimes creating a signet-ring appearance. These cells are pathognomonic for chordoma when present. The tumor grows in lobules within abundant myxoid matrix.
Q: What is the most important prognostic factor for chordoma?
A: Surgical margin status. Wide negative margins are associated with significantly better local control and overall survival. Intralesional surgery (tumor violation) results in 50-90% local recurrence at 5 years, compared to 10-30% recurrence with wide en bloc resection. En bloc resection with negative margins is the only curative treatment.
Q: Why is proton beam therapy preferred over conventional photon radiation for chordoma?
A: Chordomas are relatively radioresistant and require high radiation doses (greater than 70 Gy) for local control. These doses exceed the tolerance of surrounding normal tissues (brainstem, spinal cord, bowel) with conventional photon radiation. Proton beam therapy allows delivery of high doses to the tumor with minimal exit dose due to the Bragg peak phenomenon, thereby sparing adjacent critical structures. This achieves better tumor control with fewer complications than conventional radiation.
At a Glance
Chordoma is a malignant tumor arising from embryonic notochord remnants, accounting for 50% at the sacrum, 35% at the skull base/clivus, and 15% in the mobile spine (cervical most common). Histologically, physaliferous cells (vacuolated, bubbly cytoplasm) are pathognomonic, and brachyury is a specific nuclear immunohistochemical marker (T-box transcription factor); tumors are also S100+ and cytokeratin+. Chordoma is slow-growing but locally aggressive with high recurrence rates—wide surgical resection with clear margins is the only chance for cure, as intralesional margins result in 50-90% local recurrence. Proton beam radiotherapy is used for unresectable tumors or as adjuvant therapy. Conventional chemotherapy has limited efficacy. Ten-year survival is approximately 40-60%.
50-35-15Chordoma Location Distribution
Hook:Remember 50-35-15: Sacrum, Skull, Spine percentages decrease!
PBSChordoma Histology Features
Hook:PBS staining = Physaliferous, Brachyury, S100 for chordoma diagnosis!
WIDEChordoma Treatment Principles
Hook:Go WIDE with margins to cure chordoma - intralesional surgery fails!
Exam Viva Scenarios
Practise clinical reasoning and management decisions out loud
“A 55-year-old man presents with 12 months of progressive sacral pain and recent onset constipation. MRI shows a large midline sacral mass destroying S3-S5 with extensive anterior soft tissue extension. Biopsy shows physaliferous cells and is brachyury positive. How do you manage him?”
“A 48-year-old woman had sacral chordoma treated 3 years ago with intralesional surgery and conventional radiation. She now presents with recurrent pain. MRI shows tumor recurrence involving S2-S4. What is your approach?”
“A 60-year-old presents with a destructive midline sacral lesion on MRI that is very bright on T2 sequences. The differential diagnosis includes chordoma versus chondrosarcoma. How do you differentiate these pathologically?”
Key Facts (Must Know)
- **Notochord remnant** origin (embryonic structure, normally regresses)
- Location: **Sacrum 50%**, Skull base 35%, Mobile spine 15%
- Incidence: 1-4% of bone tumors, 0.08 per 100,000 population
- Peak age: 50-70 years, male-to-female ratio 2:1
- Slow-growing but **locally aggressive** malignancy
Histology (PBS Mnemonic)
- **P**hysaliferous cells - bubbly vacuolated cytoplasm (pathognomonic)
- **B**rachyury positive - nuclear T-box transcription factor (diagnostic)
- **S**100 and cytokeratin positive - also EMA positive
- Lobulated architecture in abundant myxoid matrix
- Brachyury distinguishes from chondrosarcoma (negative in CS)
Imaging Features
- MRI: T1 hypointense, **T2 very hyperintense** (myxoid matrix)
- CT: Midline lytic destruction with soft tissue mass
- Calcification in 30-70% (flocculent pattern)
- Heterogeneous enhancement post-contrast
- Chest CT mandatory for staging (lungs common metastatic site)
Treatment (WIDE Mnemonic)
- **W**ide margins - en bloc resection only cure
- **I**ntralesional = recurrence (50-90% recurrence rate)
- **D**ifficult locations - sacrum/skull base complex surgery
- **E**xcellent radiation - proton beam for unresectable/positive margins
- No role for conventional chemotherapy (not chemosensitive)
Sacral Surgery Pearls
- **S1 roots**: Bilateral preservation essential for walking
- **S2 roots**: Bilateral preservation for bladder/bowel function
- S3 sacrifice: Some dysfunction but better tolerated than S2
- Combined anterior-posterior approach for large tumors
- Margin status most important prognostic factor
Prognosis
- 5-year survival: 65-80%, 10-year survival: 40-60%
- Wide margins: 10-30% recurrence at 5 years
- Intralesional: 50-90% recurrence at 5 years
- Metastases occur in 10-40% (lungs, bone, liver)
- Dedifferentiated variant: Median survival 1 year
Differential Diagnosis
- **Chondrosarcoma** - brachyury negative, cytokeratin negative
- Metastatic adenocarcinoma - clinical history, brachyury negative
- Myxopapillary ependymoma - GFAP positive, no physaliferous cells
- Ecchordosis physaliphora - benign notochord remnant, small, no destruction
High-Yield Exam Points
- Brachyury is THE diagnostic marker (know this)
- En bloc wide resection is only cure (emphasize margins)
- Proton beam preferred over conventional radiation (radioresistance)
- S2 root preservation critical for bladder/bowel function
- No role for chemotherapy (but mention targeted trials)
Evidence Base
- ESMO-convened consensus of over 40 chordoma experts plus Chordoma Foundation
- En bloc resection with wide margins is the standard for resectable disease
- High-dose particle therapy (proton/carbon ion) for unresectable disease or positive margins
- Multidisciplinary, expert-centre management essential given rarity
- No standard systemic therapy; clinical trial enrolment encouraged
- Brachyury (nuclear T-box transcription factor) expressed in all 53 chordomas studied
- Negative in over 300 other neoplasms, including 163 chondroid/cartilaginous tumours
- Also negative in nucleus pulposus, arguing against direct notochordal origin of disc
- Establishes brachyury as a specific marker of notochord and notochord-derived tumours
- 29 en bloc primary sacral tumour resections; chordoma the most frequent type (16 cases)
- Wide margins in 19, marginal in 9, contaminated in 1
- Level of nerve-root transection classification predicts bladder, bowel and ambulatory function
- Median Kaplan-Meier disease-free survival for chordoma 68 months
- Margins should not be compromised to preserve function when needed for tumour control
- Prospective phase II trial: 50 patients (29 chordoma, 14 chondrosarcoma) of thoracolumbar/sacral spine
- Combined photon/proton RT to doses up to 76.6-77.4 GyRBE around resection
- 5- and 8-year local control 94% and 85% for primary tumours; 81% and 74% overall
- Recurrence 11% in primary versus 50% in previously recurrent tumours (p=0.002)
- 8-year grade 3-4 late toxicity 13%, with no radiation myelopathy
- 39 patients (30 sacral, 9 mobile spine); mean follow-up 8.1 years
- Local recurrence 44%, metastases 28%; 5-, 10-, 20-year survival 84%, 64%, 52%
- Inadequate margins, larger size, Ki-67 over 5%, tumour necrosis and recurrence were adverse factors
- Local recurrence strongly associated with metastasis and tumour-related death (p less than 0.001)
- Fine-needle aspiration favoured to avoid contamination from open biopsy
- SEER analysis of 400 microscopically confirmed chordomas, 1973-1995
- Age-adjusted incidence 0.08 per 100,000; male predominance (0.10 vs 0.06)
- Axial distribution roughly even: 32% cranial, 32.8% spinal, 29.2% sacral
- Younger and female patients more likely to have cranial disease
- Median survival 6.29 years; 5- and 10-year relative survival 67.6% and 39.9%
- Largest prospective phase II systemic trial: 56 patients with PDGFB/PDGFRB-positive advanced chordoma
- Imatinib 800 mg/day until progression
- Objective response rate only 2% (1 partial response); 70% stable disease
- 64% clinical benefit rate and median progression-free survival 9 months
- Confirms modest, mainly disease-stabilising activity in an orphan disease