Marble Bone Disease | Osteoclast Dysfunction | Dense But Brittle Bones
- Osteoclast Dysfunction: Failure of bone resorption, NOT increased bone formation
- Malignant Infantile (ARO): Life-threatening, pancytopenia, cranial nerve compression, death by age 10 without BMT
- Benign Adult (ADO): Often incidental, may present with fractures, Type II more common than Type I
- Radiographic Triad: Sandwich vertebrae, Erlenmeyer flask deformity, bone-in-bone appearance
- Surgical Challenge: Dense but brittle bone, difficult drilling, high implant failure, delayed/nonunion
- “Osteoclasts present but dysfunctional (carbonic anhydrase II or chloride channel defects)
- “Dense on X-ray but WEAK in reality - paradoxically fragile
- “Bone marrow failure from obliterated medullary canal causes pancytopenia
- “Foraminal narrowing causes optic nerve and facial nerve palsies
Bone is radiodense but mechanically weak. Osteopetrotic bone lacks normal architecture - unmineralized cartilage cores persist within bone, making it brittle like chalk. Despite appearing strong on X-ray, these patients fracture easily.
ARO without BMT is fatal by age 10. Pancytopenia from medullary obliteration, hepatosplenomegaly (extramedullary hematopoiesis), blindness from optic canal stenosis, recurrent infections. BMT is curative if done early.
Technical challenges: Drill bits break, screws strip, bone cuts difficult, tourniquet time prolonged. High failure rates: Delayed union, nonunion, and implant failure are all markedly more frequent than in normal bone. Use large drill bits, sharp instruments, patience.
Sandwich vertebrae: Sclerotic endplates with lucent center. Erlenmeyer flask: Flared metaphyses (distal femur). Bone-in-bone: Endobone appearance. Diffuse sclerosis throughout skeleton.
- ARO (Malignant Infantile)
- Autosomal recessive
- ADO Type II (Benign Adult)
- Autosomal dominant
- ARO (Malignant Infantile)
- 1:250,000
- ADO Type II (Benign Adult)
- 1:20,000 (most common form)
- ARO (Malignant Infantile)
- TCIRG1 (50%), CLCN7, OSTM1
- ADO Type II (Benign Adult)
- CLCN7 (70%)
- ARO (Malignant Infantile)
- Infancy (first year)
- ADO Type II (Benign Adult)
- Adolescence/adulthood
- ARO (Malignant Infantile)
- Fatal by age 10 without BMT
- ADO Type II (Benign Adult)
- Normal
- ARO (Malignant Infantile)
- Obliterated - pancytopenia
- ADO Type II (Benign Adult)
- Preserved - normal counts
- ARO (Malignant Infantile)
- Blindness, deafness, facial palsy common
- ADO Type II (Benign Adult)
- Rare cranial nerve issues
- ARO (Malignant Infantile)
- Marked (extramedullary hematopoiesis)
- ADO Type II (Benign Adult)
- None
- ARO (Malignant Infantile)
- Very high
- ADO Type II (Benign Adult)
- Moderate (lower limbs)
- ARO (Malignant Infantile)
- Bone marrow transplant (curative)
- ADO Type II (Benign Adult)
- Supportive, fracture management
MARBLEOsteopetrosis Features
Hook:MARBLE bone disease - dense like marble but breaks like chalk!
SEBERadiographic Features
Hook:SEBE - See Every Bone Enhanced on X-ray!
Overview and Epidemiology
Definition
Osteopetrosis (marble bone disease, Albers-Schonberg disease) is a group of rare inherited skeletal disorders characterized by defective osteoclast-mediated bone resorption. This leads to accumulation of primary spongiosa and calcified cartilage, resulting in generalized skeletal sclerosis. Despite increased radiographic density, the bone is structurally abnormal and paradoxically brittle.
Epidemiology
- Autosomal Recessive Osteopetrosis (ARO): 1 in 250,000 births - severe, infantile onset
- Autosomal Dominant Osteopetrosis (ADO): 1 in 20,000 - mild, adult onset
- Intermediate Autosomal Recessive: Variable, childhood onset
- Equal male:female ratio
- Higher incidence in consanguineous populations (ARO)
- ADO Type II is the most common form overall
Genetics
- TCIRG1 (50%): Encodes a3 subunit of vacuolar H+-ATPase (proton pump)
- CLCN7 (15%): Chloride channel 7
- OSTM1: Osteopetrosis-associated transmembrane protein
- TNFSF11 (RANKL): Osteoclast-poor form
- TNFRSF11A (RANK): Osteoclast-poor form
- CLCN7 (70%): Most common cause of ADO Type II
- LRP5: Associated with ADO Type I
CLCN7 mutations can cause both ARO and ADO - the severity depends on whether mutations are biallelic (recessive, severe) or monoallelic (dominant, mild). This explains phenotypic variability.
Pathophysiology
The Core Defect: Osteoclast Failure
Osteopetrosis is fundamentally a disease of osteoclast dysfunction - the osteoclasts are present (in most forms) but cannot resorb bone effectively.
Normal Osteoclast Function:
- Osteoclast attaches to bone surface (sealing zone)
- Proton pump (H+-ATPase) acidifies resorption lacuna
- Chloride channel (CLCN7) provides charge balance
- Acid dissolves hydroxyapatite mineral
- Cathepsin K digests collagen matrix
- Resorption products endocytosed
Pathologic Mechanisms:
- Protein
- Proton pump a3 subunit
- Function
- Acid secretion
- Result of Mutation
- Cannot acidify lacuna
- Protein
- Chloride channel 7
- Function
- Charge balance
- Result of Mutation
- Cannot maintain acid pH
- Protein
- Carbonic anhydrase II
- Function
- H+ generation
- Result of Mutation
- No protons for pump
- Protein
- CLCN7 partner
- Function
- Channel stability
- Result of Mutation
- CLCN7 degraded
- Protein
- Osteoclast formation
- Function
- Differentiation
- Result of Mutation
- Osteoclast-poor form
Consequence: No bone resorption despite normal osteoblast function leads to:
- Accumulation of calcified cartilage cores
- Primary spongiosa not converted to mature bone
- Generalized skeletal sclerosis with abnormal architecture
Clinical Presentation
Autosomal Recessive Osteopetrosis

Presentation: First year of life, often within months of birth.
- Severe pancytopenia - pallor, fatigue, failure to thrive
- Recurrent infections (pneumonia, sepsis)
- Bleeding/bruising (thrombocytopenia)
- Hepatosplenomegaly - massive, from extramedullary hematopoiesis
- Progressive blindness - optic nerve compression (50-80%)
- Deafness - auditory nerve compression
- Facial palsy - facial nerve compression
- Hydrocephalus (foramen magnum stenosis)
- Developmental delay
- Macrocephaly - skull thickening
- Frontal bossing
- Fractures (even birth trauma)
- Failure to thrive
- Delayed tooth eruption
- Dental abscesses
- Osteomyelitis of mandible (major morbidity)
- Fatal by age 10 without bone marrow transplant
- Death from infection, bleeding, or anemia
- Progressive blindness and neurological deterioration
Early diagnosis is critical. Bone marrow transplant in infancy offers the best chance of cure. In the largest international registry, 5-year survival was about 62% with an HLA-matched sibling versus about 42% with alternative donors. Delayed transplant has worse outcomes because of established, often irreversible, neurological damage.
Investigations
Blood Tests
- Full blood count: Pancytopenia in ARO, usually normal in ADO
- Blood film: Nucleated red cells, immature white cells (extramedullary hematopoiesis)
- Reticulocyte count: May be elevated (hemolysis)
- Calcium: Normal, low, or high (variable)
- Phosphate: Usually normal
- Alkaline phosphatase: May be elevated (osteoblast activity)
- Acid phosphatase (TRAP): Elevated (osteoclast marker)
- Creatine kinase BB isoenzyme: Elevated (osteoclast marker in ARO)
- PTH: May be elevated (secondary hyperparathyroidism)
- TCIRG1, CLCN7, OSTM1, TNFSF11, TNFRSF11A
- Important for prognosis and family counseling
- Determines eligibility for BMT
- Difficult aspiration (sclerotic bone)
- Trephine biopsy may show abnormal architecture
- Reduced or absent hematopoietic tissue

Differential Diagnosis
Sclerosing Bone Disorders
- Key Differentiator
- Osteoclast dysfunction, Erlenmeyer flask, sandwich vertebrae
- Genetics
- TCIRG1, CLCN7
- Key Differentiator
- Short stature, open fontanelles, acro-osteolysis, mandible hypoplasia
- Genetics
- CTSK (cathepsin K)
- Key Differentiator
- Diaphyseal involvement, limb pain, symmetric long bone sclerosis
- Genetics
- TGFB1
- Key Differentiator
- Dripping candle wax appearance, dermatomal distribution
- Genetics
- LEMD3 (mosaic)
- Key Differentiator
- Spotted bones, asymptomatic, bone islands
- Genetics
- LEMD3
- Key Differentiator
- Prostate/breast cancer history, focal lesions
- Genetics
- Acquired
Key Distinguishing Points:
- Pyknodysostosis: Toulouse-Lautrec had this; characterized by short stature, fragile bones, but with ACRO-OSTEOLYSIS (absent in osteopetrosis) and open fontanelles
- Engelmann Disease: Affects diaphyses primarily, causes pain and weakness, autosomal dominant
- Melorheostosis: Unilateral, follows sclerotome/dermatomal pattern, looks like dripping candle wax
Management
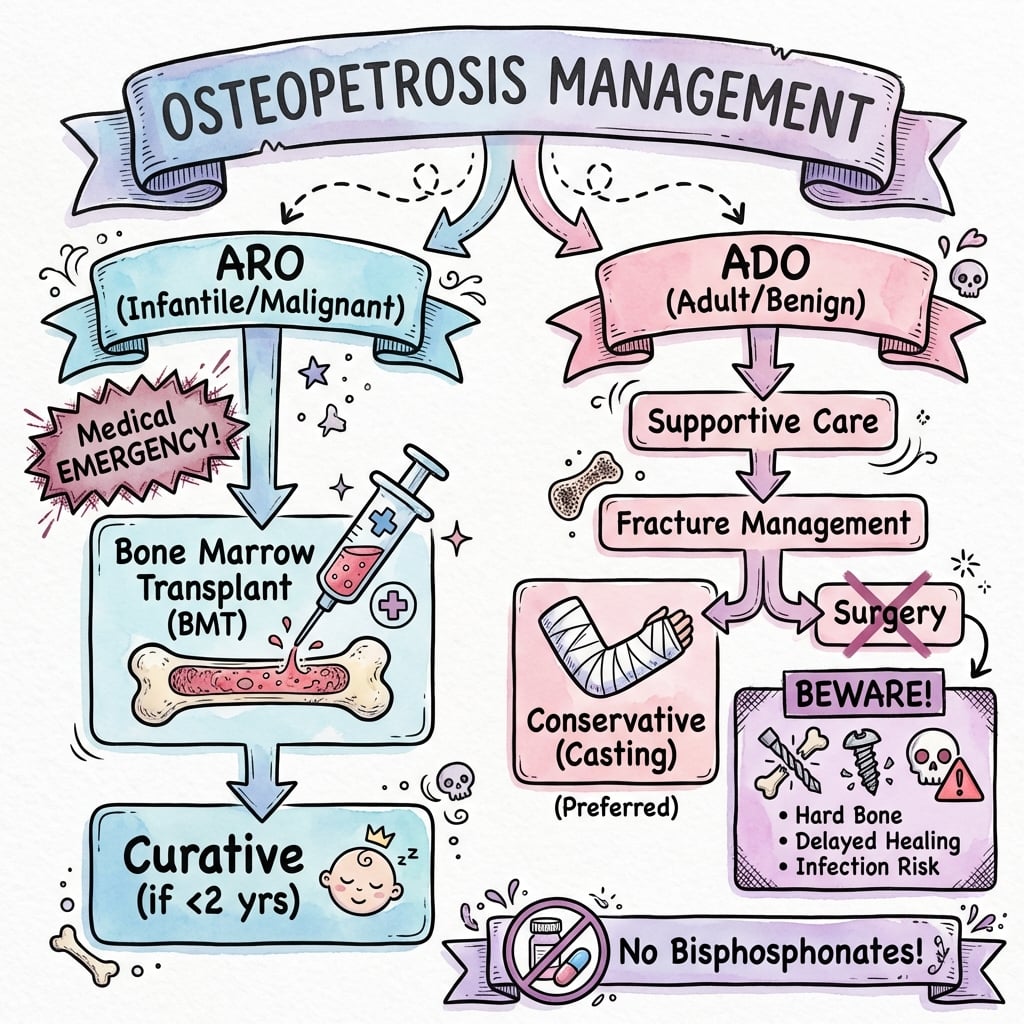
Treatment by Disease Type
- ONLY curative treatment for most forms of ARO
- Donor osteoclast precursors can form functional osteoclasts
- Best outcomes if performed early in infancy (before irreversible neurological damage)
- International registry 5-year survival: approximately 62% with an HLA-matched sibling donor
- Approximately 42% with alternative (mismatched related or unrelated) donors
- Graft failure is the leading cause of post-transplant death
- Confirmed ARO with functional osteoclast defects
- Severe hematologic involvement
- Progressive disease
- RANKL/RANK mutations (osteoclast-poor form - donor cells cannot form osteoclasts)
- Established severe neurological damage (may proceed if other organs threatened)
- Transfusions for anemia/thrombocytopenia
- Antibiotics for infections
- Vitamin D and calcium supplementation (if hypocalcemic)
- Interferon-gamma: May enhance osteoclast function
- Corticosteroids: Short-term for pancytopenia (limited role)
- No specific medical treatment - supportive care only
- Manage fractures and complications
- Optimize bone health (vitamin D, calcium)
- Avoid bisphosphonates (further impair resorption)
- Dental hygiene to prevent osteomyelitis
Do NOT give bisphosphonates in osteopetrosis. These drugs inhibit osteoclast function - the exact problem in osteopetrosis. They will worsen the disease.
Surgical Management
Orthopaedic Challenges
Technical Difficulties:
- Extremely hard cortical bone
- Drill bits break frequently (use fresh, sharp bits)
- Prolonged drilling time generates heat
- Saw blades dull rapidly
- Use larger diameter drill bits (less likely to break)
- Intermittent drilling with irrigation to prevent thermal necrosis
- Screws strip easily in abnormal bone
- Plates may not seat properly on irregular surface
- Consider locked plates (angle-stable, less reliance on bone quality)
- IM nails difficult to insert (obliterated canal)
- May need to ream canal if attempting nailing
- Delayed union very common (no bone remodeling)
- Nonunion markedly more common than in normal bone
- Callus forms but may not mature
- Fractures may heal to some degree if aligned and immobilized
- Strongly consider for non-displaced fractures
- Cast immobilization for extended periods (3-6 months)
- May achieve union with patience
- Avoids surgical complications
- Displaced fractures
- Failure of conservative treatment
- Femoral neck fractures (high nonunion risk)
- Open fractures
- Use new, sharp instruments (change drill bits frequently)
- Large diameter drills (3.2mm or larger if possible)
- Intermittent drilling with saline irrigation
- Longer screws with larger diameter if bone allows
- Locked plating preferred (less torque on screws)
- Consider external fixation (avoids screw placement in dense bone)
- Bone grafting may help (autograft preferred)
- Prolonged immobilization postoperatively

HARDSurgical Challenges
Hook:Operating on osteopetrosis is HARD - be prepared for technical challenges!
Complications
Disease-Related Complications
- ARO Complications
- Severe pancytopenia, need for transfusions, infections
- ADO Complications
- Usually none or mild anemia
- ARO Complications
- Blindness (50-80%), deafness, facial palsy, hydrocephalus
- ADO Complications
- Rare cranial nerve involvement
- ARO Complications
- Fractures, growth retardation, skeletal fragility
- ADO Complications
- Fractures (main problem), delayed healing
- ARO Complications
- Delayed eruption, abscesses, mandibular osteomyelitis
- ADO Complications
- Osteomyelitis after dental work
- ARO Complications
- Hepatosplenomegaly, extramedullary hematopoiesis
- ADO Complications
- None
Surgical Complications
- Intraoperative fracture: Brittle bone fractures during manipulation
- Drill bit breakage: Bits left in bone (usually left in situ)
- Prolonged operative time: Increased infection risk
- Bleeding: May be significant from abnormal bone
- Thermal necrosis: From prolonged drilling
- Delayed union (very common - expect as norm)
- Nonunion (markedly more common than in normal bone)
- Implant failure: Screw loosening, plate breakage
- Refracture: After implant removal or adjacent to hardware
- Infection: Increased susceptibility, difficult to eradicate
BMT Complications
- Graft-versus-host disease (GVHD)
- Graft failure
- Infection (during immunocompromised period)
- Veno-occlusive disease
- Late effects of conditioning chemotherapy
Guidelines, Registries & Global Practice
Global Epidemiology
- Figure
- approximately 1 in 250,000 births
- Source population
- Worldwide; higher in consanguineous populations
- Figure
- approximately 1 in 20,000 births
- Source population
- Most common form overall
- Figure
- approximately 70% of cases explained by 10+ genes
- Source population
- International cohorts
- Figure
- Costa Rica, Middle East, parts of Northern Europe
- Source population
- Founder and consanguinity effects
There is no high-level (RCT) guidance for this rare disease; practice rests on registry data and expert consensus.
Consensus Positions Across Societies
- Working consensus
- Allogeneic HSCT
- Comment
- Best survival with HLA-matched sibling; refer in infancy
- Working consensus
- Mandatory
- Comment
- RANKL/TNFSF11 (osteoclast-poor) forms are NOT cured by HSCT
- Working consensus
- Contraindicated as disease treatment
- Comment
- They further suppress osteoclasts; denosumab studied only for post-HSCT hypercalcaemia
- Working consensus
- Anticipate hard, brittle bone
- Comment
- Locked plating favoured; external fixation as backup
- Working consensus
- Vision, hearing, FBC, dental
- Comment
- Optic-canal decompression considered before established atrophy
Registries and Networks
HSCT outcomes are pooled through international transplant registries (CIBMTR in North America, EBMT in Europe), which together generated the largest osteopetrosis transplant cohort to date. Unrelated-donor matching relies on national and international marrow donor registries. Rare-bone-disease reference networks (for example ERN-BOND in Europe) coordinate diagnosis, genetic confirmation and multidisciplinary care.
High- vs Limited-Resource Practice Variation
- Well-resourced settings: next-generation sequencing gene panels, early HSCT in infancy, skull-base and ophthalmology services, and locked-plate plus external-fixation inventories for fracture surgery.
- Limited-resource settings: diagnosis is often clinical and radiographic; transplant access may be delayed or unavailable, shifting care toward transfusion support, infection control and conservative fracture management. Consanguinity raises ARO incidence, so genetic counselling and antenatal diagnosis carry high value where available.
Carbonic Anhydrase II Deficiency (Guibaud-Vainsel Syndrome)
The CA2 gene appears in the pathophysiology table, the intermediate-forms tab, the cheat sheet and the genetics differential, but this distinct, highly examinable syndrome is never developed - and it is the one osteopetrosis variant with a signature systemic triad.
- A distinct autosomal-recessive triad. Carbonic anhydrase II (CA2) deficiency causes a recognisable syndrome of (1) osteopetrosis + (2) renal tubular acidosis (RTA) + (3) cerebral (basal ganglia) calcification - often remembered together, with developmental delay/intellectual disability and short stature completing the picture. It is also called Guibaud-Vainsel syndrome or "marble brain disease".
- Why one enzyme does all three. Carbonic anhydrase II generates the protons (H⁺) that the osteoclast's proton pump secretes to acidify the resorption lacuna - so its loss impairs bone resorption (osteopetrosis). The same enzyme is needed for renal tubular acid handling (both proximal and distal), producing a mixed proximal + distal RTA; and it is expressed in the brain, where its deficiency is associated with the characteristic basal-ganglia calcification. One shared biochemical step, three organ systems.
- How it differs from "classic" osteopetrosis - and why it matters. Unlike TCIRG1/CLCN7 disease, CA2 deficiency is milder in its skeletal severity (marrow failure is usually NOT life-threatening), the sclerosis may even diminish with age, and crucially it is NOT curable by haematopoietic stem-cell transplant - because the defect is a systemic enzyme deficiency, HSCT corrects the marrow-derived osteoclast but not the renal or cerebral disease. Management is therefore supportive: treat the RTA (alkali therapy), manage fractures, and monitor growth/development.
Q: A child has osteopetrosis, a metabolic (renal tubular) acidosis and basal-ganglia calcification - what is the diagnosis and does BMT cure it? A: Carbonic anhydrase II (CA2) deficiency (Guibaud-Vainsel / "marble brain" syndrome) - the autosomal-recessive triad of osteopetrosis + renal tubular acidosis + cerebral calcification, often with intellectual disability and short stature. CA2 makes the protons for both the osteoclast and the renal tubule, hence one enzyme, three organs. It is milder skeletally and NOT cured by HSCT (the renal/cerebral disease persists); manage with alkali for the RTA and supportive fracture/growth care.
Osteopetrorickets: the Paradox of Rickets in Dense Bone
The Shapiro evidence card notes "thickened growth plates if there is superimposed rickets" and the pathophysiology tab lists "rickets-like features paradoxically possible", but the topic never explains how a disease of too much bone can coexist with rickets - a classic exam curveball.
- It is real, and it makes mechanistic sense. "Osteopetrorickets" is the coexistence of rachitic (unmineralised) growth-plate changes on a background of dense osteopetrotic bone - seen particularly in severe infantile ARO. The key is that osteopetrosis is a defect of resorption, not of mineral supply, and the two problems attack different compartments.
- Why the osteoclast defect causes a mineralisation failure. Normal osteoclasts liberate calcium (and phosphate) from bone into the blood; when they cannot resorb, the skeleton behaves as a calcium "sink" that traps mineral and cannot release it, so despite radiodense bones the child can become hypocalcaemic. The hypocalcaemia drives secondary hyperparathyroidism, and the low available mineral at the growth plate produces genuine rickets (widened, frayed, cupped physes) - hence dense metaphyses with rachitic growth plates side by side.
- Clinical consequences to recognise. The hypocalcaemia can be symptomatic - tetany and seizures in infancy (a feature specifically noted in ARO) - and the picture can be worsened by coexisting nutritional vitamin-D deficiency. Treatment is calcium and vitamin D/calcitriol to correct the mineral deficit (calcitriol may additionally stimulate residual osteoclasts); this is one reason mineral status is monitored closely, and it does not contradict the "no antiresorptives" rule.
Q: How can a child with osteopetrosis (dense bone) also have rickets and hypocalcaemia? A: Osteopetrosis is a defect of resorption, not mineralisation. Because osteoclasts normally release calcium from bone into the blood, a failed osteoclast makes the skeleton a calcium sink - the child becomes hypocalcaemic (with secondary hyperparathyroidism and even tetany/seizures in infancy), and the low mineral at the physis produces genuine rachitic growth plates on a dense-bone background ("osteopetrorickets"). Treat with calcium and vitamin D/calcitriol, not antiresorptives.
Controversies & Areas of Uncertainty
The rarity of osteopetrosis means most management rests on registries and expert opinion rather than randomised trials. Several genuine areas of debate persist:
-
Optimal timing and conditioning for HSCT. Earlier transplantation in infancy is associated with better outcomes, but the ideal age threshold, conditioning regimen, and donor hierarchy beyond matched siblings remain unsettled. Graft failure and hepatic/pulmonary toxicity are the dominant limitations rather than the transplant decision itself.
-
Reversibility of neurosensory deficits. Whether established optic atrophy or hearing loss can be salvaged by transplant or surgical decompression is uncertain; most evidence suggests vision rarely improves once atrophy is fixed, which is why decompression is considered only before established damage.
-
Role of optic-canal decompression. Indications and benefit are inconsistent across centres, and it is reserved for selected, progressive cases rather than offered routinely.
-
Pharmacological alternatives to transplant. Interferon-gamma-1b offers only modest, mainly supportive benefit; recombinant RANKL for osteoclast-poor (TNFSF11) disease and gene therapy for TCIRG1/CLCN7 remain investigational, with limited human data.
-
Denosumab in osteopetrosis. As a disease therapy it is contraindicated (it suppresses osteoclasts), yet it has been explored narrowly for transplant-related hypercalcaemia - a nuance that is easy to misstate.
-
Fracture fixation strategy. There is no consensus trial comparing plating, intramedullary fixation, and external fixation; choice is individualised, and the often-quoted precise nonunion percentages are extrapolated from small series rather than robust cohort data.
Self-Assessment Questions
What is the fundamental defect in osteopetrosis?
A: Osteoclast dysfunction leading to failure of bone resorption. The osteoclasts are present (in most forms) but cannot resorb bone effectively due to defects in proton pump (TCIRG1), chloride channel (CLCN7), or carbonic anhydrase (CA2). This leads to accumulation of calcified cartilage and primary spongiosa.
Which gene is most commonly mutated in autosomal recessive osteopetrosis?
A: TCIRG1 (50% of ARO cases). This gene encodes the a3 subunit of the vacuolar H+-ATPase (proton pump) essential for acidification of the resorption lacuna.
Name three classic radiographic features of osteopetrosis.
A: Sandwich vertebrae (rugger jersey spine - sclerotic endplates with lucent center), Erlenmeyer flask deformity (flared metaphyses from failed tubulation), and bone-in-bone appearance (endobone phenomenon).
Why is bone marrow transplant curative for most forms of ARO?
A: Osteoclasts are derived from hematopoietic stem cells. Donor HSCs can differentiate into functional osteoclasts that restore bone resorption. However, RANKL-deficient forms do NOT respond because osteoclast precursors cannot differentiate without RANKL signal.
Why are delayed union and nonunion common after fractures in osteopetrosis?
A: Fracture healing depends on coordinated bone remodelling, which requires functional osteoclasts. In osteopetrosis the osteoclast defect abolishes normal remodelling, so callus forms but cannot be reorganised into mature lamellar bone. The result is a markedly elevated rate of delayed union and nonunion compared with normal bone, alongside the technical difficulty of fixation in dense, brittle bone.
Why are bisphosphonates absolutely contraindicated in osteopetrosis?
A: Bisphosphonates inhibit osteoclast function - the exact problem in osteopetrosis. Administration would further impair bone resorption and worsen the disease.
Viva Scenarios
Practise clinical reasoning and management decisions out loud
“You are asked to see a 6-month-old infant diagnosed with autosomal recessive osteopetrosis. The child has pancytopenia, hepatosplenomegaly, and progressive vision loss. How would you manage this patient?”
“A 35-year-old man with known benign adult osteopetrosis (ADO Type II) presents with a displaced mid-shaft femoral fracture after a fall. How would you manage this fracture?”
“You are shown X-rays of the lumbar spine and distal femur showing diffuse sclerosis, 'sandwich vertebrae', and flaring of the distal femoral metaphysis. What is the diagnosis and what are the key radiographic features of this condition?”
PATHOPHYSIOLOGY
- Osteoclast DYSFUNCTION (not absent)
- Failed bone RESORPTION
- Dense but BRITTLE bone
- Medullary canal OBLITERATED
TYPES
- ARO: Autosomal recessive, infantile, FATAL without BMT
- ADO Type II: Most common, benign, adult, CLCN7
- ADO Type I: Rare, cranial vault involvement
- Intermediate: Variable, childhood onset
RADIOGRAPHIC FEATURES
- Sandwich vertebrae (rugger jersey)
- Erlenmeyer flask (flared metaphyses)
- Bone-in-bone (endobone)
- Diffuse osteosclerosis
ARO COMPLICATIONS
- Pancytopenia (marrow obliteration)
- Blindness (optic canal stenosis)
- Hepatosplenomegaly (extramedullary hematopoiesis)
- Death by age 10 without BMT
GENES
- TCIRG1: 50% of ARO (proton pump)
- CLCN7: ARO and ADO (chloride channel)
- RANKL: Osteoclast-poor (no BMT benefit)
- CA2: With renal tubular acidosis
SURGICAL CHALLENGES
- Drill bits BREAK - use large diameter, change often
- Screws STRIP - use locked plates
- Healing DELAYED - high delayed-union and nonunion risk
- Consider EXTERNAL FIXATION as alternative
TREATMENT
- ARO: BMT curative (early, before age 2)
- ADO: Supportive, fracture management
- NO bisphosphonates (worsen disease)
- Interferon-gamma (bridge therapy)
Evidence Base
- Authoritative review of osteopetrosis pathophysiology and classification
- Osteoclast development vs function defects distinguished as the unifying mechanism
- Proton pump (TCIRG1), chloride channel (CLCN7) and RANK/RANKL pathways mapped to phenotypes
- Haematopoietic stem cell transplantation framed as the only cure for osteoclast-rich infantile disease
- ARO incidence 1 in 250,000 births; ADO incidence 1 in 20,000 births
- At least 10 causative genes account for roughly 70% of all cases
- Severe infantile forms cause death in the first decade if untreated; adult-onset forms have normal life expectancy
- Diagnosis is clinical and radiographic, confirmed by gene testing where applicable