Vaso-occlusion and Bone Complications
- HbS: Glutamate to valine substitution at position 6 of beta-globin chain
- Vaso-occlusion: Sickling under hypoxia/acidosis causes bone infarcts, AVN, painful crises
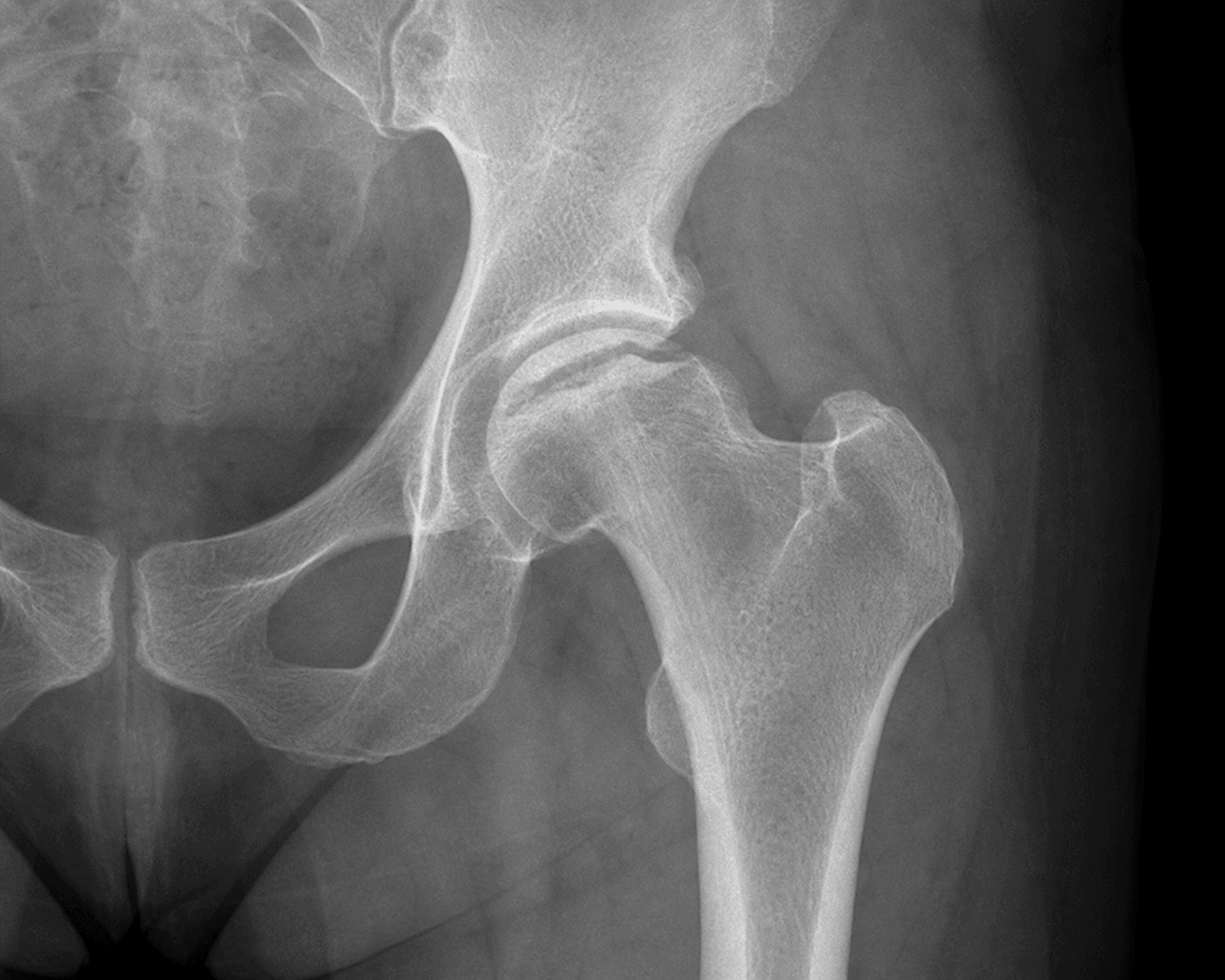
- AVN: Femoral head (10-30%), humeral head - often bilateral
- Osteomyelitis: SALMONELLA is most common organism (unique to SCD, unlike general population)
- Perioperative: Avoid hypothermia, hypoxia, dehydration, acidosis - transfuse HbS to less than 30%
- “Salmonella osteomyelitis
- “H-shaped vertebrae
- “Bilateral AVN common
- “Hydration critical perioperatively
In Sickle Cell Disease, the most common osteomyelitis organism is SALMONELLA.
- This is UNIQUE to SCD - viva examiners will test this
- Staph aureus is still common but Salmonella is MORE common
- Empiric antibiotics MUST cover BOTH organisms
- Autosplenectomy increases infection susceptibility
Overview and Epidemiology
Sickle Cell Disease (SCD) is a hereditary hemoglobinopathy with significant orthopaedic manifestations.
- Inheritance: Autosomal recessive
- Mutation: HBB gene - Glutamate to Valine substitution at position 6 (Glu6Val)
- Genotypes: HbSS (most severe), HbSC (milder), HbS-beta thalassemia
- Birth burden: Over 300,000 affected births annually worldwide; roughly 75-80% occur in sub-Saharan Africa, with India and the Middle East also high-burden
- Carrier protection: HbS trait confers resistance to falciparum malaria (balanced polymorphism), explaining its distribution across the historical malaria belt
- Migration: Prevalence rising in Europe, North America and Australasia through migration; diaspora populations now make SCD a global, not regional, orthopaedic problem
- Survival: Median survival 40-60+ years in high-resource settings with hydroxyurea, vaccination and transfusion programmes; remains markedly lower in limited-resource regions where childhood mortality is high
Pathophysiology and Mechanisms
- HbS Formation: Single nucleotide mutation in beta-globin gene (Glu6Val)
- Deoxygenation triggers polymerization: HbS molecules aggregate when deoxygenated
- Polymer formation: Long polymer fibres distort red cell membrane into sickle shape
- Membrane damage: Repeated sickling causes irreversible membrane damage
- Sickled cells are rigid and adhere to endothelium
- Obstruction of microvasculature (capillaries, sinusoids)
- Ischemia of downstream tissues
- Reperfusion injury with inflammatory response
- Marrow infarction: Leads to painful crises and bone necrosis
- End-artery occlusion: Particularly affects epiphyses (AVN) and endplates (H-vertebrae)
- Marrow hyperplasia: Compensatory expansion to combat chronic haemolysis
- Cortical thinning: Secondary to marrow expansion
- Increased infection risk: Functional asplenia + micro-infarcts create nidus
- Cold exposure
- Hypoxia
- Acidosis
- Infection
- Dehydration
Clinical Presentation
Painful Vaso-occlusive Crisis
- Severe bone pain - often diaphyseal
- Dactylitis (hand-foot syndrome) in children less than 3 years
- Long bone pain in older children and adults
- No fever or mild low-grade temperature
- Dehydration
- Infection
- Cold exposure
- High altitude/hypoxia
- Physical exertion
- Swelling and tenderness over affected bones
- No fluctuance or abscess
- Often multiple sites
- Duration typically 4-7 days
Vaso-occlusive crisis is the most common acute complication requiring hospitalisation.
Investigations
Laboratory Investigations
- Full blood count: Chronic anaemia (Hb 6-9 g/dL typical)
- Reticulocyte count: Elevated (chronic haemolysis)
- Blood film: Sickled cells, Howell-Jolly bodies (asplenia)
- Haemoglobin electrophoresis: Confirms HbSS, HbSC, or HbS-thal
- Blood cultures (if febrile)
- CRP, ESR: Elevated in infection
- Procalcitonin: May help differentiate crisis from infection
- WCC: Very elevated suggests infection vs mild elevation in crisis
- Group and hold/crossmatch
- Coagulation studies
- Renal function (may have sickle nephropathy)
- Liver function (iron overload from transfusions)
HbS percentage is critical for surgical planning - target less than 30%.
Differentiating Crisis vs Osteomyelitis:
- Vaso-occlusive Crisis
- Acute (hours)
- Osteomyelitis
- Subacute (days to weeks)
- Vaso-occlusive Crisis
- Low-grade or absent
- Osteomyelitis
- High-grade (38.5C or higher)
- Vaso-occlusive Crisis
- Normal or mildly elevated
- Osteomyelitis
- Significantly elevated
- Vaso-occlusive Crisis
- Mildly elevated (under 100)
- Osteomyelitis
- Markedly elevated (over 100)
- Vaso-occlusive Crisis
- Often multifocal
- Osteomyelitis
- Usually single focus
- Vaso-occlusive Crisis
- Marrow oedema only
- Osteomyelitis
- Periosteal reaction, soft tissue changes, abscess
- Vaso-occlusive Crisis
- 4-7 days typical
- Osteomyelitis
- Weeks if untreated
- Vaso-occlusive Crisis
- Good improvement
- Osteomyelitis
- Minimal improvement
Differential Diagnosis of the Painful Limb/Joint in SCD:
- Distinguishing Features
- Multifocal, symmetrical, recurrent, low-grade or no fever, responds to hydration/analgesia
- Key Investigation
- MRI: marrow oedema, no periosteal reaction
- Distinguishing Features
- Single focus, high fever, persistent despite hydration, soft-tissue signs
- Key Investigation
- MRI plus blood/bone cultures (Salmonella greater than Staph)
- Distinguishing Features
- Hot, swollen joint, refusal to move, often hip in children with AVN
- Key Investigation
- Joint aspiration (urgent)
- Distinguishing Features
- Insidious groin/shoulder pain, mechanical, reduced internal rotation, often bilateral
- Key Investigation
- MRI (earliest), staged by Ficat/ARCO
- Distinguishing Features
- Chest/back pain, fever, hypoxia, new infiltrate - may mimic rib infarct
- Key Investigation
- CXR plus SpO2; high mortality
- Distinguishing Features
- Monoarticular, hyperuricaemia from cell turnover, crystals
- Key Investigation
- Joint aspiration for crystals
Management
Vaso-occlusive Crisis Management
- Hydration: IV fluids at 1.5x maintenance
- Analgesia: Often requires opioids (morphine PCA)
- Oxygen: If SpO2 less than 95%
- Warmth: Maintain normothermia
- Transfusion: Simple transfusion if severe
- Rest
- NSAIDs (caution with renal function)
- Antiemetics if needed
- Monitor for complications
- Acute chest syndrome (fever, respiratory symptoms, new infiltrate)
- Severe anaemia (Hb less than 5 g/dL)
- Stroke symptoms
- Priapism lasting more than 4 hours
Most crises resolve within 4-7 days with supportive care.
Complications
- Avascular necrosis: 10-50% develop AVN of femoral head, humeral head
- Pathological fractures: Weakened bone prone to fracture
- Chronic osteomyelitis: Recurrent or incompletely treated infections
- Growth disturbance: Limb length discrepancy, angular deformity
- Joint contractures: From repeated crises and immobilisation
- Infection: 10-15% (vs 1-2% general population)
- Sickling crisis: Perioperative trigger by hypoxia/hypothermia
- Acute chest syndrome: Postoperative complication, potentially life-threatening
- Wound healing: Delayed healing common
- Implant failure: Earlier loosening and revision
- Acute chest syndrome: Most common cause of death in SCD
- Stroke: Occurs in 10% of children with SCD
- Renal failure: Sickle nephropathy
- Pulmonary hypertension: From chronic haemolysis
- Iron overload: From chronic transfusions
Acute Chest Syndrome: the Perioperative Killer
The topic repeatedly names acute chest syndrome (ACS) - as the single most common cause of death in SCD, as an escalation criterion in vaso-occlusive crisis, and as a postoperative complication that incentive spirometry is used to prevent - but the orthopaedic surgeon must understand it in its own right, because it is the commonest life-threatening perioperative event in these patients. In the PTSCD surgical trial ACS occurred in 10% of patients in both transfusion arms and caused the only two deaths, so it is not abolished by transfusion alone.
ACS is a new pulmonary infiltrate on chest imaging involving at least one complete lung segment, plus a new respiratory sign or symptom - fever, cough, chest or back pain, tachypnoea, wheeze or hypoxia. It is clinically and radiologically indistinguishable from pneumonia at onset, so the two are managed together.
- Fat and marrow embolism from infarcted long-bone marrow (bone-marrow necrosis showers fat emboli to the lungs) - relevant after fracture, marrow-cavity instrumentation or a severe long-bone crisis.
- Hypoventilation and atelectasis from post-operative pain, opioid respiratory depression, and splinting after thoracic, upper-abdominal, rib or sternal incisions/infarcts.
- Immobility, over-transfusion/fluid overload, and infection in the perioperative period.
new or worsening hypoxia, tachypnoea, chest/back pain, fever and a new infiltrate on chest radiograph. Distinguish from pneumonia, pulmonary embolism, fluid overload and (in the trauma patient) classic fat-embolism syndrome - though in SCD these mechanisms overlap.
escalate early to HDU/ICU. Give supplemental oxygen titrated to saturations, incentive spirometry, bronchodilators if wheeze, and broad antibiotics covering atypical and encapsulated organisms. Provide analgesia that is adequate to prevent splinting yet titrated to avoid opioid-induced hypoventilation. Simple transfusion improves oxygen-carrying capacity, and exchange transfusion is indicated for severe or deteriorating hypoxia. Prevention is the surgeon's lever: incentive spirometry, early mobilisation, judicious (not excessive) fluids, and regional analgesia to limit systemic opioids, layered on the normothermia-oxygenation-transfusion protocol.
Post-operative hypoxia in a sickle-cell patient is acute chest syndrome until proven otherwise. It complicated roughly 1 in 10 operations in the PTSCD trial and was the only cause of death in that study, so transfusing to Hb 10 g/dL does not remove the risk. The orthopaedic-specific triggers are fat/marrow embolism from infarcted marrow and opioid-related hypoventilation with splinting - which is exactly why incentive spirometry, early mobilisation and opioid-sparing regional analgesia sit in the perioperative protocol. Treat with oxygen, antibiotics covering atypicals, and simple or (if severe) exchange transfusion.
Dactylitis (Hand-Foot Syndrome): Often the First Manifestation
Dactylitis is named in the clinical presentation and MCQ points as the "hand-foot syndrome" of infancy but is never developed, yet it is frequently the earliest skeletal manifestation of SCD and the event that first brings the child to the diagnosis.
vaso-occlusive infarction of the marrow of the small tubular bones - the metacarpals, metatarsals and phalanges. It clusters in infants and children younger than 3 years of age, the period before the red (haematopoietic) marrow of the hands and feet converts to yellow marrow; as that conversion completes, dactylitis largely disappears and crises shift to the long bones and axial skeleton.
- Painful, diffuse dorsal swelling of the hands and/or feet, warmth and tenderness, often with a low-grade fever and reluctance to use the limb.
- Characteristically bilateral, symmetrical and multi-digit - a pattern that helps separate it from a single-focus infection.
films are normal or show only soft-tissue swelling in the first days; after roughly one to two weeks, periosteal reaction, patchy medullary lucency and sclerosis appear from the infarct, occasionally a bone-within-bone appearance. Changes usually resolve without deformity, but rare epiphyseal/physeal damage can leave a shortened digit (brachydactyly) or metacarpal/metatarsal length discrepancy.
osteomyelitis or septic arthritis (single focus, higher and persistent fever, unresponsive to hydration), non-accidental injury in the swollen infant, and - in tuberculosis-endemic regions - tuberculous dactylitis (spina ventosa). Management is the same as any vaso-occlusive crisis: hydration, analgesia, warmth and reassurance, as the episode is self-limiting over one to two weeks.
Symmetrical, painful swelling of the hands and feet in an infant can be the presenting feature of sickle cell disease. Dactylitis is infarction of the metacarpal, metatarsal and phalangeal marrow and is confined to the under-3 age group because it depends on the red marrow that later converts to yellow. Its bilateral, multi-digit pattern and self-limiting course distinguish it from osteomyelitis (single focus, persistent high fever) - but always consider non-accidental injury and, in endemic areas, tuberculous spina ventosa. Treat as a vaso-occlusive crisis; radiographic periosteal/medullary changes lag the pain by one to two weeks.
Outcomes
- THA survivorship: 80-90% at 10 years (lower than general population)
- Infection rate: 10-15%
- Dislocation rate: Increased
- Revision rate: Higher due to loosening and infection
- HbSS genotype: Worse prognosis than HbSC
- Frequent crises: Associated with more complications
- Age: Earlier AVN associated with worse long-term outcomes
- Transfusion burden: Iron overload complications
- Modern care: Median survival 40-60 years
- Hydroxyurea: Reduces crisis frequency, improves survival
- Stem cell transplant: Potentially curative in selected patients
- SCD
- 80-90%
- General Population
- 95-98%
- SCD
- 10-15%
- General Population
- 1-2%
- SCD
- Increased
- General Population
- 1-3%
- SCD
- Earlier failure
- General Population
- 1-2% at 10 years
- SCD
- Higher
- General Population
- Standard
Guidelines, Registries & Global Practice
Global epidemiology:
- Over 300,000 affected births per year worldwide; the great majority in sub-Saharan Africa, with India and the Middle East also high-burden.
- Diaspora migration has made SCD a routine consideration in orthopaedic units across Europe, North America and Australasia.
- Femoral head osteonecrosis affects roughly 10% (higher with age and HbSS + alpha-thalassaemia); osteomyelitis and septic arthritis are over-represented compared with the general population.
Side-by-side society guidance:
- Position
- Preoperative simple transfusion to Hb approximately 10 g/dL for most surgery under GA; extended antigen matching to limit alloimmunisation
- Position
- Multidisciplinary haematology-led perioperative planning; hydroxycarbamide (hydroxyurea) for recurrent crises; transfusion to reduce HbS for high-risk surgery
- Position
- Hydroxyurea for frequent crises/acute chest syndrome; chronic transfusion for primary stroke prevention; penicillin prophylaxis and full vaccination in children
- Position
- No SCD-specific implant mandate; emphasis on thermoregulation, oxygenation, hydration, controlled transfusion and anticipating sclerotic/obliterated canals
- No dedicated SCD arthroplasty registry exists; national joint registries (NJR, AOANJRR, AJRR, Swedish/Norwegian) capture osteonecrosis as an indication but rarely sub-stratify SCD, so most outcome data come from single-centre and pooled series (e.g. the Hernigou experience).
- High-resource: newborn screening, hydroxyurea, vaccination, antigen-matched blood, MRI access, planned exchange transfusion and elective protocol-driven arthroplasty.
- Limited-resource: late presentation, restricted safe-blood and MRI availability, higher childhood mortality, and a greater burden of advanced osteonecrosis and chronic osteomyelitis presenting for salvage rather than elective reconstruction.
- Universal principles regardless of setting: haematology co-management, avoidance of the sickling triggers, infection coverage for Salmonella plus Staph, and informed counselling about elevated surgical risk.
Controversies and Areas of Uncertainty
- Transfusion strategy for major orthopaedic surgery: The PTSCD trial validated simple transfusion to Hb 10 g/dL for most surgery, but whether the highest-risk arthroplasties (long operative time, HbSS, prior acute chest syndrome) still benefit from exchange to HbS under 30% remains unsettled and is individualised with haematology.
- Tourniquet use: Traditionally avoided for fear of regional hypoxia/acidosis precipitating sickling, but several series report safe use with limited time and good oxygenation. There is no high-level evidence either way - practice varies.
- Core decompression and biologics: Outcomes of core decompression (with or without concentrated bone marrow / cell therapy) are less predictable than in non-SCD osteonecrosis; patient selection (pre-collapse, small lesions) is key and the evidence base is largely observational.
- Cemented vs cementless fixation: Abnormal sclerotic bone and obliterated medullary canals complicate both techniques. No consensus exists; choice is guided by bone quality, canal anatomy and surgeon experience rather than robust comparative data.
- Distinguishing infarct from osteomyelitis: Even MRI plus inflammatory markers can be equivocal; some advocate early aspiration/biopsy, others a trial of conservative management with close monitoring. Over-diagnosis leads to unnecessary surgery; under-diagnosis risks sepsis.
- Curative therapies: Allogeneic stem cell transplant and emerging gene therapies may alter the future orthopaedic disease burden, but their long-term effect on established bone disease is unknown.
MCQ Practice Points
Q: What is the most common cause of osteomyelitis in patients with sickle cell disease?
A: Salmonella species is the most common organism causing osteomyelitis in sickle cell disease (approximately 50%), unlike the general population where S. aureus predominates. Reason: Splenic dysfunction (autosplenectomy) impairs clearance of encapsulated organisms and Salmonella. S. aureus is still the second most common. Clinical challenge: Differentiating bone infarction (vaso-occlusive crisis) from osteomyelitis - both present with fever, pain, and elevated inflammatory markers. MRI helps differentiate (osteomyelitis shows soft tissue abscess, cortical destruction).
Q: What are the orthopaedic manifestations of sickle cell disease?
A: Avascular necrosis: Femoral head (most common - 10-30% of patients), humeral head, vertebral bodies; due to vaso-occlusive crisis affecting end-arterial blood supply. Osteomyelitis: Increased risk, Salmonella most common pathogen. Bone infarcts: Long bone diaphyses; may mimic osteomyelitis. Dactylitis ("hand-foot syndrome"): Painful swelling of hands/feet in infants - first manifestation of SCD. Growth disturbance: Vertebral end-plate collapse ("H-shaped" or "Lincoln log" vertebrae). Pathological fractures from weakened bone.
Q: How do you differentiate bone infarction from osteomyelitis in sickle cell disease?
A: Clinical overlap: Both cause pain, fever, swelling, elevated WBC/CRP/ESR. Favoring osteomyelitis: Localized warmth, erythema, soft tissue abscess, single bone involvement, persistent fever despite hydration/analgesia. Favoring infarction: Multiple bone involvement, symmetric, responds to hydration/pain management. Imaging: MRI - osteomyelitis shows soft tissue collection, cortical destruction, enhancing abscess; infarction shows serpentine enhancement pattern. Aspiration/biopsy: Definitive - culture positive in osteomyelitis. When in doubt, treat empirically for osteomyelitis covering Salmonella and S. aureus.
Q: What perioperative considerations are important in patients with sickle cell disease undergoing orthopaedic surgery?
A: Preoperative: Hematology consultation; consider preoperative transfusion to achieve HbS less than 30% and Hb 10g/dL (exchange transfusion if needed); optimize hydration. Intraoperative: Avoid hypoxia, acidosis, hypothermia, dehydration (all precipitate sickling); use supplemental oxygen; warm IV fluids; careful tourniquet use (controversial - limit time, ensure adequate oxygenation). Postoperative: Continue supplemental oxygen; aggressive pain management; early mobilization; incentive spirometry (prevent acute chest syndrome); maintain hydration. Higher risk of VTE, infection, and wound complications.
Q: What are the treatment options for avascular necrosis of the femoral head in sickle cell disease?
A: Treatment mirrors AVN from other causes but with specific considerations: Early stages (Ficat I-II): Protected weight-bearing, core decompression (mixed results in SCD). Advanced stages (Ficat III-IV): Total hip arthroplasty - higher complication rate in SCD (infection, wound problems, perioperative crisis) but outcomes improving with modern perioperative protocols. Specific considerations: Younger patient age often; higher revision rates than non-SCD; cement may be preferred (abnormal bone quality); aggressive perioperative transfusion reduces complications. Preoperative optimization critical.
Self-Assessment Quiz
At a Glance
Sickle cell disease (SCD) is an autosomal recessive hemoglobinopathy caused by the HbS mutation (glutamate to valine at position 6). Sickling of red cells under hypoxia causes vaso-occlusion leading to bone infarcts, painful crises, and avascular necrosis (10-50% prevalence, commonly bilateral femoral head). The critical exam point is that Salmonella is the most common osteomyelitis organism in SCD (unique to this condition), requiring dual antibiotic coverage. Perioperative management must avoid the "5 Hs" that trigger sickling: Hypoxia, Hypothermia, Hypovolemia (dehydration), Hydrogen ions (acidosis), and Hypotension. Preoperative exchange transfusion targets HbS under 30%.
SCDSCD Orthopaedic Complications
Hook:SICKLE - the shape that causes the disease reminds you of all complications.
CHANTPerioperative Protocol: CHANT
Hook:CHANT the mantra to avoid sickling crisis.
HAIRRadiographic Signs
Hook:HAIR on end skull appearance from marrow expansion.
Viva Scenarios
Practise clinical reasoning and management decisions out loud
“10-year-old with known sickle cell disease (HbSS) presents with fever 39C, right tibial pain and swelling for 1 week. Mum says it's different from his usual pain crises.”
“Same patient now 30 years old requires total hip arthroplasty for Ficat Stage IV AVN of right hip. How do you prepare him for surgery?”
“22-year-old woman with HbSS sickle cell disease presents with 3-month history of bilateral hip pain and limp. X-rays show early sclerosis of both femoral heads without collapse.”
“You are shown a lateral spine X-ray of a 15-year-old with sickle cell disease showing characteristic central endplate depression of multiple vertebral bodies. Describe the findings and explain the pathophysiology.”
PATHOPHYSIOLOGY
- HbS: Glu6Val mutation
- Sickling under hypoxia/acidosis
- Vaso-occlusion causes infarcts
- Autosomal recessive
ORTHOPAEDIC ISSUES
- AVN: Hip/shoulder, 10-50%
- Osteomyelitis: SALMONELLA before Staph
- Bone crises: Painful vaso-occlusion
- H-shaped vertebrae: Endplate infarcts
PERIOPERATIVE (CHANT)
- Cold avoidance
- Hydration critical
- Acidosis prevention
- Normal oxygen (SpO2 95%+)
- Transfuse: HbS less than 30%, Hb 10
THA OUTCOMES
- Infection: 10-15%
- Survivorship: 80-90% at 10yr
- Higher revision rates
- Counsel about complications
Evidence Base
- Multicentre RCT, 604 operations randomised to aggressive (target HbS less than 30%) vs conservative (target Hb 10 g/dL) transfusion
- Serious complications similar between groups (31% vs 35%)
- Aggressive arm had double the transfusion-related complications (14% vs 7%)
- Acute chest syndrome in 10% of both groups
- 2590 patients followed mean 5.6 years; femoral head osteonecrosis prevalence approximately 10% at entry
- Highest incidence in HbSS with co-inherited alpha-thalassaemia (4.5 cases/100 patient-years)
- Frequent painful crises and higher haematocrit associated with osteonecrosis
- Hip arthroplasty results poor - 5 of 27 needed reoperation within 11-53 months
- 2524 patients; humeral head osteonecrosis prevalence 5.6% at entry
- Highest incidence in HbSS with alpha-thalassaemia and S/beta-zero-thalassaemia
- Only 20.9% had pain or restricted movement at diagnosis (often silent)
- Frequent in children and young adults
- 22-year review; 10 osteomyelitis and 4 septic arthritis cases in children with SCD
- Salmonella was the offending organism in 8 of 10 osteomyelitis cases
- Plain films and bone scans were unreliable for separating infection from infarct
- Aspiration/biopsy recommended in the ill, febrile (greater than 38.2C) child
- Double-blind RCT, 299 adults with 3 or more crises/year
- Hydroxyurea reduced median crises from 4.5 to 2.5 per year (p less than 0.001)
- Fewer acute chest syndrome episodes (25 vs 51) and fewer transfusions (48 vs 73)
- No important short-term adverse effects
- Synthesis of 2126 single-surgeon SCD THAs over 40 years plus 3742 from the literature
- Provides structured perioperative recommendations to reduce the high complication burden
- Emphasises haematology co-management, controlled transfusion and meticulous thermoregulation/oxygenation
- Acknowledges higher infection, loosening and revision rates than primary OA
- Preoperative simple transfusion to Hb approximately 10 g/dL advised for most patients undergoing surgery with general anaesthesia
- Extended red-cell antigen matching to limit alloimmunisation
- Aggressive exchange not routinely superior to simple transfusion for moderate-risk surgery
- Individualise in chronically transfused or highly alloimmunised patients