Lysosomal Storage Disorders | Dysostosis Multiplex | Spinal Instability
- Dysostosis multiplex - pathognomonic radiographic pattern: J-shaped sella, paddle ribs, anterior vertebral beaking, bullet-shaped metacarpals
- Cervical instability - atlantoaxial subluxation due to odontoid hypoplasia, most severe in MPS IV (Morquio)
- Anaesthetic risks - difficult airway, atlantoaxial instability, restrictive lung disease require specialist input
- MPS IV (Morquio) - unique because intelligence is PRESERVED but skeletal manifestations most severe
- Enzyme replacement therapy - available for MPS I, II, IVA, VI but does NOT cross blood-brain barrier
- “MPS IV (Morquio) has NORMAL intelligence but most severe skeletal disease
- “Odontoid hypoplasia causes atlantoaxial instability - high anaesthetic risk
- “Carpal tunnel syndrome common in MPS - often first presenting sign in adults
- “HSCT must be done before age 2 for cognitive benefit in MPS I
All MPS patients have potential atlantoaxial instability due to odontoid hypoplasia and ligamentous laxity. ALWAYS assess cervical spine before any procedure. Flexion-extension views required. High anaesthetic mortality without precautions.
Difficult intubation due to: short neck, large tongue, mandibular hypoplasia, tonsillar hypertrophy, tracheal narrowing from GAG deposition. Require experienced paediatric anaesthetist and fibreoptic intubation available.
Only MPS with normal intelligence but most severe skeletal manifestations. Universal odontoid hypoplasia (50% symptomatic cord compression), severe genu valgum, thoracolumbar kyphosis. Major surgical candidate.
Classic radiographic pattern in all MPS types: J-shaped sella turcica, paddle ribs, anterior vertebral beaking, proximal metacarpal pointing, flared iliac wings. First thing to look for on X-rays.
Overview and Epidemiology
The mucopolysaccharidoses (MPS) are a group of inherited lysosomal storage disorders caused by deficiency of enzymes required for the degradation of glycosaminoglycans (GAGs), formerly called mucopolysaccharides. Accumulation of GAGs in tissues leads to progressive multisystem disease with prominent skeletal manifestations. [1]
GAGs are complex carbohydrates that form essential components of connective tissue matrix, including dermatan sulfate, heparan sulfate, keratan sulfate, and chondroitin sulfate. In MPS, deficient lysosomal enzymes cannot break down specific GAGs, leading to accumulation in lysosomes. This causes cellular dysfunction, inflammation, and progressive tissue damage. [2]
The combined incidence of all MPS types is approximately 1:25,000 live births, though this varies by population and type. [1,3]
- MPS I (Hurler/Scheie): 1:100,000
- MPS II (Hunter): 1:100,000-150,000 (X-linked, males only)
- MPS III (Sanfilippo): 1:70,000 (most common)
- MPS IV (Morquio): 1:75,000-100,000
- MPS VI (Maroteaux-Lamy): 1:250,000
- MPS VII (Sly): Very rare, fewer than 1:250,000
A growing number of jurisdictions now include MPS I in newborn screening programs. Early diagnosis enables HSCT before irreversible neurological damage occurs. The critical window for transplantation is before age 2 years for cognitive benefit.
Pathophysiology and Genetics
Glycosaminoglycan Metabolism: Glycosaminoglycans are long, unbranched polysaccharide chains composed of repeating disaccharide units. They are attached to core proteins to form proteoglycans, which are essential components of extracellular matrix, particularly in cartilage, bone, and connective tissue.
The Four Main GAGs:
- Location
- Skin, blood vessels, heart valves
- MPS Types Affected
- MPS I, II, VI, VII
- Location
- CNS, liver, retina
- MPS Types Affected
- MPS I, II, III, VII
- Location
- Cornea, cartilage, intervertebral discs
- MPS Types Affected
- MPS IV
- Location
- Cartilage, bone, heart
- MPS Types Affected
- MPS VII
Pathogenesis of Skeletal Disease:
- Chondrocyte dysfunction - GAG accumulation impairs normal chondrocyte maturation
- Growth plate disorganization - disrupted columnar arrangement reduces longitudinal growth
- Abnormal endochondral ossification - leads to short stature and limb deformities
- Bone matrix abnormalities - irregular mineralization causes osteopenia
- Ligamentous laxity - accumulated GAGs weaken collagen cross-linking
In MPS, odontoid hypoplasia results from defective endochondral ossification. Combined with ligamentous laxity from GAG accumulation in periarticular tissues, this creates atlantoaxial instability. The posterior arch of C1 moves anteriorly relative to C2, compressing the cervical cord. This is most severe in MPS IV (Morquio syndrome).
A key examinable subtlety: cervical cord compression in MPS is frequently driven as much by anterior soft-tissue GAG deposition as by the bony odontoid hypoplasia, and this explains the patient who is myelopathic despite a near-normal atlantodental interval:
- Dural / ligamentous thickening (dural dysplasia): GAG accumulates in the periodontoid ligaments, the posterior longitudinal ligament and the dura, producing a thickened retro-odontoid soft-tissue mass ("pseudotumour") and circumferential canal narrowing that compresses the cord anteriorly.
- Consequence for imaging: an MPS patient can have significant myelopathic stenosis with only a modest ADI - so the space available for the cord (SAC) and the MRI appearance of the soft tissues matter more than the ADI alone. Always look at the cord and the soft tissue, not just the bony measurement.
- Consequence for surgery: a posterior fusion alone may not relieve an anterior soft-tissue compression. Many MPS patients need a decompression (posterior, or occasionally anterior/transoral for a large retro-odontoid mass) combined with fusion. Encouragingly, the retro-odontoid soft-tissue mass can regress after stabilisation, and disease-modifying therapy may reduce ongoing GAG deposition.
Exam point: in MPS, cord compression is bony (odontoid hypoplasia) plus soft-tissue (dural/ligamentous GAG); judge the cord on MRI and SAC, and remember that fusion alone may not decompress an anterior soft-tissue mass.
- Autosomal recessive: MPS I, III, IV, VI, VII (all except Hunter)
- X-linked recessive: MPS II (Hunter syndrome) - males affected, females are carriers
For autosomal recessive MPS, carrier parents have a 25% risk of affected offspring with each pregnancy. Prenatal diagnosis is available via CVS or amniocentesis for all MPS types. Preimplantation genetic diagnosis is an option for affected families.
Classification
Classification by Enzyme Deficiency:
- Eponym
- Hurler
- Deficient Enzyme
- Alpha-L-iduronidase
- GAG Stored
- DS, HS
- Intelligence
- Impaired
- Key Features
- Most severe, death by 10y
- Eponym
- Scheie
- Deficient Enzyme
- Alpha-L-iduronidase
- GAG Stored
- DS, HS
- Intelligence
- Normal
- Key Features
- Mildest, normal lifespan
- Eponym
- Hurler-Scheie
- Deficient Enzyme
- Alpha-L-iduronidase
- GAG Stored
- DS, HS
- Intelligence
- Variable
- Key Features
- Intermediate phenotype
- Eponym
- Hunter
- Deficient Enzyme
- Iduronate-2-sulfatase
- GAG Stored
- DS, HS
- Intelligence
- Variable
- Key Features
- X-linked, no corneal clouding
- Eponym
- Sanfilippo
- Deficient Enzyme
- Various
- GAG Stored
- HS
- Intelligence
- Severely impaired
- Key Features
- Severe CNS, mild skeletal
- Eponym
- Morquio A
- Deficient Enzyme
- GALNS
- GAG Stored
- KS, CS
- Intelligence
- Normal
- Key Features
- Most severe skeletal disease
- Eponym
- Morquio B
- Deficient Enzyme
- Beta-galactosidase
- GAG Stored
- KS
- Intelligence
- Normal
- Key Features
- Milder than IVA
- Eponym
- Maroteaux-Lamy
- Deficient Enzyme
- Arylsulfatase B
- GAG Stored
- DS
- Intelligence
- Normal
- Key Features
- Corneal clouding, cardiac
- Eponym
- Sly
- Deficient Enzyme
- Beta-glucuronidase
- GAG Stored
- DS, HS, CS
- Intelligence
- Variable
- Key Features
- Very rare, hydrops fetalis
DS = dermatan sulfate; HS = heparan sulfate; KS = keratan sulfate; CS = chondroitin sulfate
Clinical Presentation
Presentation age correlates with disease severity:
- Severe MPS I (Hurler): First year of life - hepatosplenomegaly, developmental delay
- Intermediate types: 2-6 years - coarse facies, joint stiffness, hernias
- Attenuated types: Childhood-adulthood - carpal tunnel, joint problems, cardiac
- Coarse facies (gargoylism) - thickened features, broad nose, thick lips
- Macrocephaly with frontal bossing
- Corneal clouding (NOT in Hunter syndrome)
- Chronic rhinorrhea and otitis media
- Hearing loss (conductive and sensorineural)
- Cognitive impairment (not in MPS IV, VI, some II)
- Communicating hydrocephalus
- Spinal cord compression (cervical, thoracolumbar)
- Carpal tunnel syndrome
- Valve thickening and regurgitation
- Coronary artery disease
- Cardiomyopathy
- Restrictive lung disease
- Obstructive sleep apnea
- Tracheobronchomalacia
Before ANY procedure in MPS patients:
- Cervical spine - flexion-extension X-rays, MRI if symptoms
- Cardiac - echocardiogram (valve disease, cardiomyopathy)
- Respiratory - pulmonary function tests, sleep study if snoring
- Airway - ENT assessment, fibreoptic equipment available
- Anaesthesia - experienced paediatric/specialist anaesthetist
- Odontoid hypoplasia (universal in MPS IV, common in others)
- Atlantoaxial instability
- Cervical stenosis
- Gibbus deformity at craniocervical junction
- Kyphosis/kyphoscoliosis
- Thoracolumbar gibbus (L1/L2 vertebral hypoplasia)
- Spinal stenosis
- Carpal tunnel syndrome (common, often bilateral)
- Trigger fingers
- Joint stiffness and contractures
- Claw hand deformity
- Restricted shoulder motion
- Genu valgum (especially MPS IV)
- Hip dysplasia and coxa valga
- Pes planus
- Ankle valgus
- Bullet-shaped or pointed proximal metacarpals (radiographic)
- Short, broad hands
- Contractures (except MPS IV which has laxity)
Investigations
- Initial screening test
- Quantitative total GAG and qualitative GAG pattern
- Elevated in most MPS, but can be normal in attenuated forms
- Pattern suggests MPS type (dermatan, heparan, keratan sulfate)
- Gold standard for diagnosis
- Performed on leukocytes, fibroblasts, or dried blood spots
- Each MPS type has specific enzyme deficiency
- Confirms diagnosis and allows carrier testing
- Identifies specific mutations
- Enables prenatal diagnosis and family screening
- Genotype-phenotype correlation possible for some types
- Radiographic Finding
- J-shaped sella turcica, thick calvarium
- Clinical Significance
- Hydrocephalus risk, increased ICP
- Radiographic Finding
- Anterior vertebral beaking (inferior), platyspondyly
- Clinical Significance
- Kyphosis, gibbus deformity
- Radiographic Finding
- Paddle or oar-shaped (widened anteriorly)
- Clinical Significance
- Restrictive lung disease
- Radiographic Finding
- Flared iliac wings, shallow acetabulum
- Clinical Significance
- Hip dysplasia, subluxation risk
- Radiographic Finding
- Widened diaphyses, coarse trabeculation
- Clinical Significance
- Fracture risk, deformity
- Radiographic Finding
- Bullet-shaped proximal metacarpals
- Clinical Significance
- Pathognomonic finding
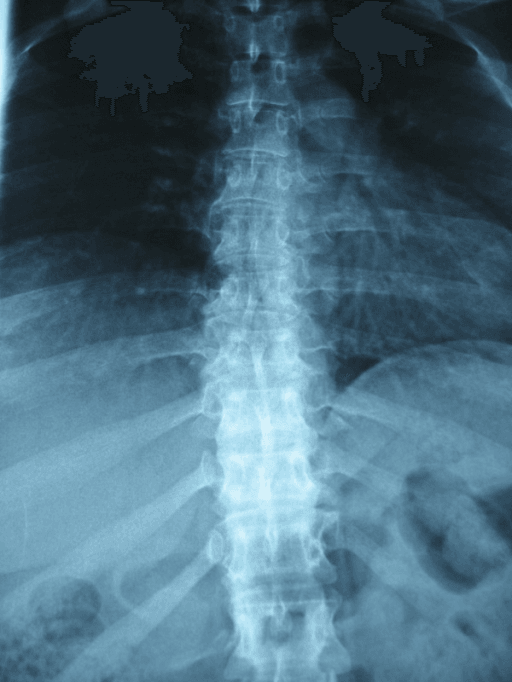
"Anterior vertebral beaking" is one of the dysostosis-multiplex hallmarks, but the location of the beak on the vertebral body at the thoracolumbar junction (the apex of the gibbus) is a classic ISAWE discriminator between the two most-tested MPS phenotypes:
- Antero-inferior (inferior) beak: the beak projects from the lower-anterior corner of the body, with hypoplasia/wedging of the antero-superior portion. This is the pattern of MPS I (Hurler) and the more severe MPS types and underlies the anterior gibbus they develop.
- Central / middle (anterior-central) beak: the beak projects from the mid-anterior vertebral body and the bodies are flattened (platyspondyly with a tongue-like central beak). This is the pattern of MPS IV (Morquio) and tracks with its universal platyspondyly and odontoid hypoplasia.
The mechanistic teaching is that the beak reflects where the body is deficient: an inferior beak goes with anterosuperior hypoplasia and a focal kyphos (Hurler), whereas a central beak goes with generalised platyspondyly (Morquio).
Exam point: on a lateral thoracolumbar film, an inferior (antero-inferior) vertebral beak suggests Hurler/severe MPS, whereas a central (mid-body) beak with platyspondyly suggests Morquio (MPS IV) - then confirm with urine GAG (keratan sulfaturia in Morquio) and the enzyme assay.
- Lateral (neutral, flexion, extension)
- AP and open mouth odontoid views
- Measure atlantodental interval (ADI): greater than 5mm concerning in children
- Gold standard for cord compression assessment
- Indicated if ADI greater than 5mm or neurological symptoms
- Shows soft tissue GAG deposition causing stenosis
- Best for bony anatomy - odontoid hypoplasia, os odontoideum
- 3D reconstructions helpful for surgical planning
Key measurements in MPS cervical spine assessment:
- Atlantodental interval (ADI): Normal less than 3mm adult, less than 5mm child. Greater than 5mm indicates instability.
- Space available for cord (SAC): Measured from posterior C1 ring to anterior C2 body. Less than 14mm indicates significant stenosis.
- Powers ratio: BC/OA. Greater than 1.0 indicates anterior atlantoaxial subluxation.
Additional Investigations:
- Indication
- All MPS patients
- Key Findings
- Valve thickening, regurgitation
- Indication
- Snoring, apnea
- Key Findings
- Obstructive sleep apnea
- Indication
- Preoperative
- Key Findings
- Restrictive pattern
- Indication
- Numbness, weakness
- Key Findings
- Carpal tunnel syndrome
- Indication
- All patients
- Key Findings
- Conductive/sensorineural loss
- Indication
- Visual symptoms
- Key Findings
- Corneal clouding, retinopathy
Differential Diagnosis:
The combination of coarse facies, short stature, joint disease and dysostosis multiplex overlaps with several other storage and skeletal dysplasias. Key discriminators are corneal clouding, intelligence, urine GAG/oligosaccharide pattern and the specific enzyme assay.
- Distinguishing features
- Coarse facies and dysostosis but NORMAL urine GAG; very high plasma lysosomal enzymes
- Skeletal pattern
- Dysostosis multiplex (resembles Hurler)
- Confirmatory test
- Plasma lysosomal enzyme activity markedly elevated; normal urine GAG
- Distinguishing features
- Coarse facies, cherry-red spot, severe neurodegeneration
- Skeletal pattern
- Dysostosis multiplex
- Confirmatory test
- Beta-galactosidase deficiency (overlaps MPS IVB)
- Distinguishing features
- Short stature and epiphyseal change WITHOUT visceromegaly, corneal clouding or raised urine GAG
- Skeletal pattern
- Epiphyseal dysplasia, no dysostosis multiplex
- Confirmatory test
- Normal urine GAG; COMP / COL2A1 / other gene testing
- Distinguishing features
- Coarse features, developmental delay, delayed bone age — reversible
- Skeletal pattern
- Epiphyseal dysgenesis, delayed ossification
- Confirmatory test
- Thyroid function tests
- Distinguishing features
- Morquio (MPS IVA/B): NORMAL intelligence, keratan sulfaturia, odontoid hypoplasia, ligamentous laxity (not stiffness)
- Skeletal pattern
- Platyspondyly, universal odontoid hypoplasia, genu valgum
- Confirmatory test
- Urine keratan sulfate; GALNS or beta-galactosidase assay
- Distinguishing features
- Inflammatory joint stiffness can mimic MPS contractures
- Skeletal pattern
- Erosive arthropathy, no dysostosis multiplex
- Confirmatory test
- Inflammatory markers, autoantibodies; normal urine GAG
Management
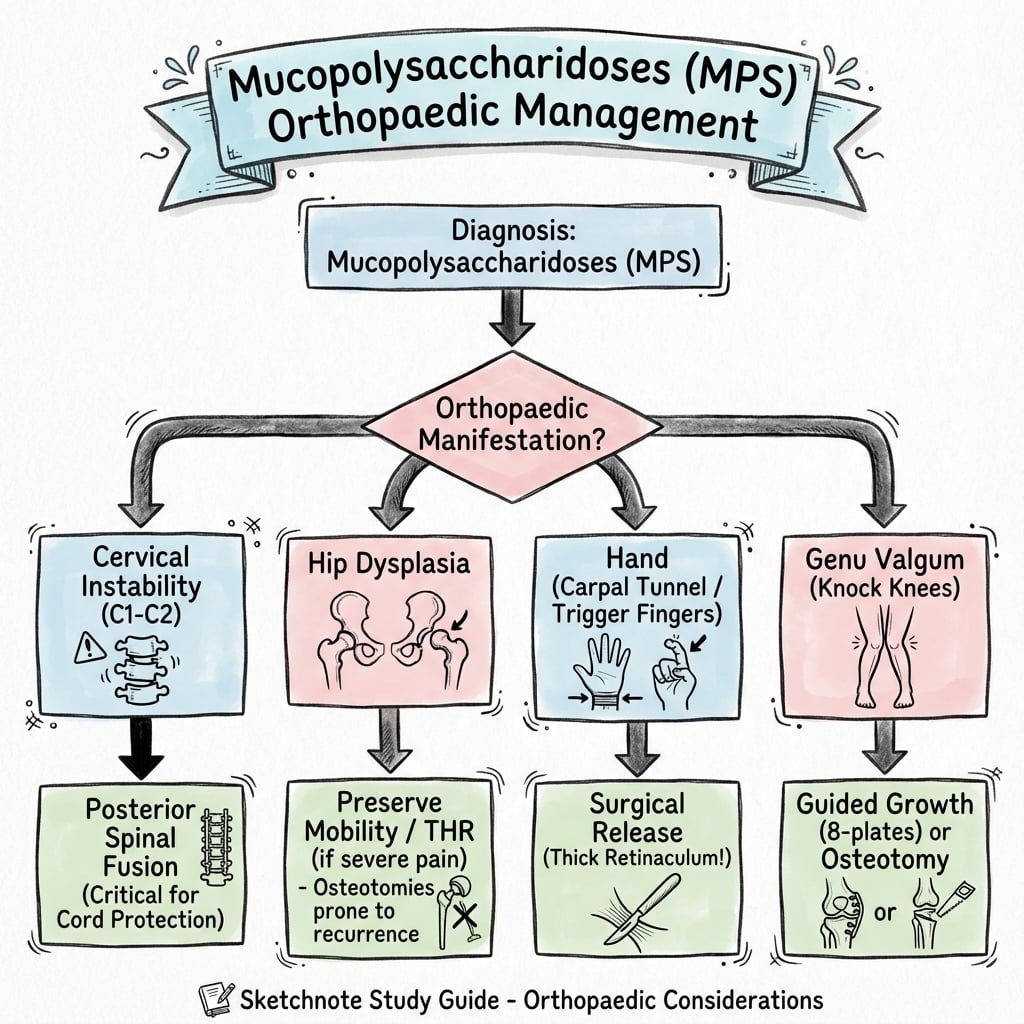
Disease-Modifying Therapies:
1. Enzyme Replacement Therapy (ERT): Available for MPS I, II, IVA, VI
- Enzyme
- Laronidase
- Brand Name
- Aldurazyme
- Dosing
- 0.58 mg/kg weekly IV
- Limitations
- Does not cross BBB
- Enzyme
- Idursulfase
- Brand Name
- Elaprase
- Dosing
- 0.5 mg/kg weekly IV
- Limitations
- Does not cross BBB
- Enzyme
- Elosulfase alfa
- Brand Name
- Vimizim
- Dosing
- 2 mg/kg weekly IV
- Limitations
- Primarily skeletal benefit
- Enzyme
- Galsulfase
- Brand Name
- Naglazyme
- Dosing
- 1 mg/kg weekly IV
- Limitations
- Significant antibody formation
- Improves respiratory function and endurance
- Reduces hepatosplenomegaly
- Some improvement in cardiac function
- Does NOT reverse established skeletal disease
- Does NOT cross blood-brain barrier (no cognitive benefit)
- Only treatment to halt cognitive decline in MPS I
- Most effective if performed before age 2 years
- Donor-derived enzyme crosses BBB
- Carries significant transplant-related mortality (10-15%)
- Standard of care for severe MPS I (Hurler)
For MPS I (Hurler), HSCT should be performed before age 2 years and ideally before DQ falls below 70. After age 2, irreversible neurological damage limits benefit. International guidelines recommend early HSCT with busulfan-based conditioning.
- Genistein (isoflavone) reduces GAG synthesis
- Investigational, may have role as adjunct therapy
- Currently in clinical trials for several MPS types
- Potential for single-treatment disease modification
- AAV-mediated liver-directed therapy most advanced
- ENT: Adenotonsillectomy for airway obstruction
- Respiratory: CPAP/BiPAP for sleep apnea
- Cardiac: Valve surgery if severe regurgitation
- Ophthalmology: Corneal transplant if severe clouding
Multidisciplinary care coordination is essential for optimal outcomes in MPS patients.
Complications
Cervical Myelopathy:
- Most serious orthopaedic complication
- Results from atlantoaxial instability and/or stenosis
- Presents with weakness, hyperreflexia, gait disturbance
- May be sudden (trauma) or insidious
- Requires urgent surgical decompression and fusion
Anaesthetic Complications:
- Mechanism
- Macroglossia, short neck, mandibular hypoplasia
- Prevention
- Fibreoptic intubation, awake technique
- Mechanism
- Atlantoaxial instability during positioning
- Prevention
- Preoperative imaging, in-line stabilization, halo
- Mechanism
- Restrictive lung disease, tracheal narrowing
- Prevention
- Preoperative PFTs, postoperative ICU
- Mechanism
- Valve disease, cardiomyopathy
- Prevention
- Preoperative echo, cardiac optimization
- Nonunion/pseudarthrosis (higher in poor bone quality)
- Hardware failure
- Neurological deterioration (positioning, decompression)
- Adjacent segment degeneration
- Recurrence of deformity (especially genu valgum)
- Hardware prominence
- Wound healing issues
- Need for repeat procedures
- Carpal tunnel recurrence
- Incomplete release
- Trigger finger recurrence
- ERT does not reverse established skeletal disease
- Skeletal manifestations continue to progress
- Multiple surgical procedures often required throughout life
- Mobility decline despite intervention
Leading causes of death in MPS:
- Respiratory failure - restrictive lung disease, sleep apnea, recurrent infections
- Cardiac failure - valve disease, cardiomyopathy
- Cervical myelopathy - sudden death from cord compression Early ERT/HSCT and proactive orthopaedic management have improved survival but do not fully normalize life expectancy.
Outcomes and Prognosis
Natural History Without Treatment:
- Untreated Life Expectancy
- 10-15 years
- Major Causes of Death
- Cardiac, respiratory
- Untreated Life Expectancy
- Normal
- Major Causes of Death
- Cardiac complications
- Untreated Life Expectancy
- 15-25 years
- Major Causes of Death
- Respiratory, cardiac
- Untreated Life Expectancy
- 15-30 years
- Major Causes of Death
- Neurological decline
- Untreated Life Expectancy
- 20-40 years
- Major Causes of Death
- Cervical myelopathy, respiratory
- Untreated Life Expectancy
- 20-40 years
- Major Causes of Death
- Cardiac, respiratory
- Survival greater than 90% with current protocols
- Cognitive stabilization if transplanted early (before age 2)
- Somatic disease continues to progress (skeletal, cardiac)
- 10-15 year post-transplant data shows improved survival
- 6-minute walk test improvement
- Reduced hepatosplenomegaly
- Some pulmonary function improvement
- Limited skeletal benefit - does not reverse deformity
- High fusion rates (greater than 90%) but technical challenges
- Neurological stabilization in most
- Revision rates 10-20% for hardware issues
- Guided growth effective but recurrence common
- Osteotomy provides definitive correction at maturity
- Improves ambulation and quality of life
- Good outcomes, symptom relief in 90%
- Recurrence rate 5-10%
- Specialized MPS clinics improve outcomes
- Coordinated medical, surgical, and supportive care
- Regular surveillance prevents complications
Guidelines, Registries & Global Practice
Global Epidemiology:
Combined birth incidence of all MPS types varies markedly by country and ascertainment method. According to PubMed, the US National MPS Society database (789 patients over 20 years) found an overall MPS birth incidence of 0.98 per 100,000 live births and prevalence of 2.67 per million, with MPS I, II and III having the highest birth incidence (0.26 per 100,000 each) — figures lower than several European series, reflecting under-ascertainment in large, fragmented populations (Puckett et al. Orphanet J Rare Dis 2021, PMID 34051828, DOI). European newborn-screening and registry studies typically report combined incidences of roughly 1 in 25,000 to 1 in 30,000, so the "1 in 25,000" figure quoted in textbooks reflects higher-ascertainment cohorts rather than a single global truth.
International Guidance — Side by Side:
There is no single orthopaedic college guideline for MPS; care is driven by international expert-consensus statements and disease registries. The table contrasts the major sources relevant to orthopaedic practice.
- Scope
- MPS I diagnosis, surveillance, HSCT vs ERT
- Key orthopaedic guidance
- Baseline multisystem assessment + review every 6-12 months; HSCT before age 2 for severe phenotype; lifelong musculoskeletal monitoring
- Evidence basis
- International expert consensus (Level V)
- Scope
- Cervical cord compression natural history
- Key orthopaedic guidance
- Routine cervical MRI from diagnosis; decompress before fixed myelopathy; anticipate high anaesthetic risk
- Evidence basis
- Prospective/retrospective registry (Level III)
- Scope
- MPS IVA natural history
- Key orthopaedic guidance
- Serial 6MWT, respiratory and cervical surveillance; document progressive functional decline
- Evidence basis
- Multinational cohort (Level II)
- Scope
- Limb and spine deformity
- Key orthopaedic guidance
- Guided growth before osteotomy for genu valgum; staged correction; multidisciplinary anaesthetic planning
- Evidence basis
- Expert/observational, no high-level RCT
The defining therapeutic evidence is consistent worldwide: weekly elosulfase alfa improves endurance in MPS IVA (Hendriksz, phase 3 RCT) and early HSCT preserves cognition in MPS I-Hurler (Aldenhoven international cohort), but neither reverses established skeletal disease — so orthopaedic surveillance and surgery remain central everywhere regardless of access to disease-modifying therapy.
Registry Evidence:
International disease registries (the MPS VI Clinical Surveillance Programme, the International Morquio Registry / MorCAP, and national MPS Society databases) provide the bulk of natural-history and surgical-outcome data, because randomised surgical trials are impossible in such rare disease. They consistently show near-universal cervical cord compression risk in MPS IVA and VI and progressive multisystem decline.
Practice Variation:
- Newborn screening: MPS I is screened in a growing number of regional newborn-screening programmes, enabling pre-symptomatic HSCT; most countries still rely on clinical diagnosis, delaying the critical pre-age-2 transplant window.
- Access to ERT: Reimbursement differs by jurisdiction. In many high-income settings, enzyme replacement therapy for MPS I (laronidase), II (idursulfase), IVA (elosulfase alfa) and VI (galsulfase) is publicly funded or reimbursed through national high-cost drug programmes; many low- and middle-income countries have no funded access, shifting management entirely onto surveillance and surgery.
- Surgical thresholds: Centres vary in whether borderline atlantoaxial instability is fused prophylactically before major surgery or monitored, reflecting the absence of randomised data.
- Model of care: Specialist multidisciplinary MPS care is delivered through paediatric tertiary centres with metabolic, anaesthetic and genetics input, paediatric bone-marrow-transplant services, and access to international haematopoietic-cell donor registries; orthopaedic procedures (cervical and thoracolumbar fusion, carpal tunnel release, guided growth and corrective osteotomy) are performed at these centres, with patient advocacy and care coordination supported by national MPS societies in many countries.
MCQ Practice Points
A: MPS IV (Morquio syndrome). This is because keratan sulfate (the stored GAG in Morquio) is predominantly found in cartilage, not brain tissue. These patients have universal odontoid hypoplasia and severe genu valgum but can participate fully in surgical decision-making.
A: MPS II (Hunter syndrome). All other MPS types are autosomal recessive. Hunter syndrome also uniquely does NOT have corneal clouding, distinguishing it from other MPS types.
A: JARS PB mnemonic: J-shaped sella turcica, Anterior vertebral beaking, Ribs paddle-shaped, Short metacarpals with proximal pointing, Pelvis with flared iliac wings, Bones with widened diaphyses.
A: Before age 2 years. Transplantation after this age will not halt or reverse cognitive decline because irreversible neurological damage has already occurred. The critical window is ideally before DQ falls below 70.
A: ERT does not cross the blood-brain barrier and cannot reach already-formed skeletal tissue with established GAG deposits. It can prevent further accumulation but cannot reverse existing damage. HSCT produces donor-derived enzyme that does cross the BBB.
At a Glance
Mucopolysaccharidoses (MPS) are lysosomal storage disorders with glycosaminoglycan accumulation causing progressive skeletal abnormalities. The hallmark radiographic pattern is dysostosis multiplex: J-shaped sella, paddle ribs, anterior vertebral beaking, and bullet-shaped metacarpals. MPS IV (Morquio syndrome) is unique—normal intelligence but most severe skeletal disease, with 50% having symptomatic atlantoaxial instability due to odontoid hypoplasia. All MPS patients carry significant anaesthetic risks (difficult airway, cervical instability, restrictive lung disease). Enzyme replacement therapy is available for MPS I, II, IVA, and VI but does not cross the blood-brain barrier; HSCT must be performed before age 2 for cognitive benefit in MPS I.
MPSMPS Types by Features - HHMMSS
Hook:HHMMSS - like counting seconds, count the MPS types. Double letters for the key ones
JARSDysostosis Multiplex X-ray Features - JARS PB
Hook:JARS PB - GAGs stored in JARS cause PB (peanut butter) thick bones
MPSOrthopaedic MPS Complications - STICK
Hook:Things STICK in MPS - GAGs stick in tissues causing all these problems
Clinical Decision Scenarios
Practise clinical reasoning and management decisions out loud
“A 12-year-old boy with Morquio syndrome (MPS IV) presents with progressive leg weakness over 6 months. He has a shuffling gait and increased tone in both legs with upgoing plantars. How would you assess and manage this patient?”
“A 3-year-old child is referred by a paediatrician with a new diagnosis of MPS I (Hurler syndrome). They want orthopaedic input on the skeletal manifestations. What are the key orthopaedic concerns and how would you structure management?”
“An 8-year-old girl with MPS IV (Morquio syndrome) is referred with progressive knock-knees. Mechanical axis shows 20 degrees of valgus bilaterally with the deformity predominantly at the distal femur. She is ambulatory but fatigues quickly. How would you manage this?”
“A 25-year-old man with MPS II (Hunter syndrome, attenuated type) presents with bilateral hand weakness. He works in IT and has noticed difficulty typing. He has normal intelligence and was diagnosed at age 15. Nerve conduction studies show severe bilateral carpal tunnel syndrome. How would you manage this?”
MPS Types - Key Features
- MPS I (Hurler) - most severe, cognitive decline, HSCT needed early
- MPS II (Hunter) - X-linked, no corneal clouding, variable severity
- MPS III (Sanfilippo) - severe CNS disease, minimal skeletal
- MPS IV (Morquio) - NORMAL intelligence, WORST skeletal disease
- MPS VI (Maroteaux-Lamy) - normal intelligence, cardiac valve disease
Dysostosis Multiplex (JARS PB)
- J-shaped sella turcica
- Anterior (inferior) vertebral beaking
- Ribs - paddle or oar shaped
- Short metacarpals with proximal pointing (bullet-shaped)
- Pelvis - flared iliac wings, shallow acetabulum
- Bones - widened diaphyses
Cervical Spine Assessment
- ADI greater than 5mm = instability (child)
- SAC less than 14mm = significant stenosis
- Odontoid hypoplasia UNIVERSAL in MPS IV
- Flexion-extension views in ALL MPS patients
- MRI if ADI elevated or neurological symptoms
Surgical Priorities
- Cervical fusion if ADI greater than 5mm or myelopathy
- Genu valgum - guided growth in growing child
- Carpal tunnel release - extended, open technique
- Always assess cervical spine before ANY surgery
- Specialist anaesthesia mandatory
Treatment Essentials
- HSCT for MPS I before age 2 for cognitive benefit
- ERT does NOT cross BBB or reverse skeletal disease
- ERT available for MPS I, II, IVA, VI
- Multidisciplinary care improves outcomes
- Regular surveillance prevents complications
Evidence Base
Enzyme Replacement Therapy for MPS IVA (landmark phase 3 RCT)
- Weekly elosulfase alfa improved 6MWT by an estimated 22.5m vs placebo at 24 weeks (P=0.017)
- Every-other-week regimen gave no significant 6MWT benefit (0.5m, P=0.954)
- Urinary keratan sulfate reduced with both active regimens
- No significant improvement in 3-minute stair-climb test
Long-Term Outcome of HSCT for MPS I (Hurler) — landmark international cohort
- 217 engrafted MPS I-H patients; median follow-up age 9.2 years
- Younger age at HSCT and preserved pre-transplant cognition predict better neurodevelopment
- Noncarrier donor with complete chimerism (normal enzyme level) gives best multisystem outcome
- Residual skeletal and cardiac disease persists in most patients despite successful HSCT
Cervical Cord Compression in MPS — international surveillance registry
- Cervical cord compression in 75.4% of MPS VI subjects with cervical MRI
- Most compression already present at first MRI; near-universal by age 20
- Surgical decompression in 42% (mean age 13.1y); reoperation in 13.8%
- Both perioperative deaths were anaesthesia-related — high airway risk
Hemiepiphysiodesis for Genu Valgum in MPS IVA (Morquio)
- Mean intermalleolar distance reduced by 6.12cm (P=0.0001)
- Mean 6-minute walk distance improved by +69.5m (P=0.034)
- No implant failure, loosening or infection; low complication rate
- Repeat plating needed in 3 of 23 patients — recurrence/staged correction expected
Morquio A Clinical Assessment Program (MorCAP) — landmark natural-history cohort
- 325 MPS IVA subjects, mean age 14.5 years; mean height z-score -5.6 (extreme short stature)
- Mean 6-minute walk distance only 212.6m, demonstrating severe endurance limitation
- Restrictive respiratory impairment (mean FVC 1.2L), worse with age
- Higher urinary keratan sulfate correlated with greater clinical impairment
International Management & Treatment Guidelines for MPS I
- Comprehensive baseline multisystem assessment mandatory for every MPS I patient
- Serial review every 6 to 12 months by a multidisciplinary team
- HSCT preferred for severe (Hurler) phenotype diagnosed early; ERT for attenuated disease
- Age, phenotype and developmental quotient drive the HSCT-versus-ERT decision
References
-
Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, et al. (eds). The Metabolic and Molecular Bases of Inherited Disease. 8th ed. McGraw-Hill; 2001:3421-3452.
-
Clarke LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology. 2011;50 Suppl 5:v13-18.
-
Puckett Y, Mallorga-Hernández A, Montaño AM. Epidemiology of mucopolysaccharidoses (MPS) in United States: challenges and opportunities. Orphanet J Rare Dis. 2021;16(1):241.
-
Lachman RS, et al. Mucopolysaccharidosis IVA (Morquio A Syndrome) and VI (Maroteaux-Lamy Syndrome): Under-recognized and Challenging to Diagnose. Insights Imaging. 2014;5(5):577-592.
-
White KK, Harmatz P. Orthopedic management of mucopolysaccharide disease. J Pediatr Rehabil Med. 2010;3(1):47-56.
-
Solanki GA, et al. Cervical cord compression in mucopolysaccharidosis VI (MPS VI): findings from the MPS VI Clinical Surveillance Program (CSP). Mol Genet Metab. 2016;118(4):310-318.
-
Hendriksz CJ, et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J Inherit Metab Dis. 2014;37(6):979-990.
-
Aldenhoven M, et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125(13):2164-2172.
-
White KK, et al. Analysis of a national MPS IVA registry: clinical characteristics, surgical interventions, and outcomes. J Bone Joint Surg Am. 2019;101(14):1256-1265.
-
Harmatz P, et al. The Morquio A Clinical Assessment Program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab. 2013;109(1):54-61.