Benign Fibro-Osseous Lesion | GNAS Mutation | Ground-Glass Appearance | Shepherd's Crook Deformity
- GNAS mutation at Arg201 codon causes constitutive cAMP activation in bone
- Ground-glass appearance on X-ray with endosteal scalloping and loss of corticomedullary differentiation
- Shepherd's crook deformity of proximal femur from repetitive microfractures in polyostotic disease
- McCune-Albright syndrome: polyostotic fibrous dysplasia plus café-au-lait macules plus endocrinopathy
- Chinese letters pattern on histology - irregular woven bone trabeculae without osteoblastic rimming
- “Fibrous dysplasia is a developmental disorder, not a true neoplasm
- “Monostotic 20:1 more common than polyostotic in most series
- “Malignant transformation under 1% (higher with radiation therapy)
- “Bisphosphonates (pamidronate, zoledronic acid) reduce bone pain and fracture risk
Postzygotic somatic mutation in GNAS gene (Arg201 codon) causes constitutive activation of Gsα protein, leading to excess cAMP production. This drives abnormal osteoblast differentiation and replacement of normal bone with fibrous tissue and immature woven bone. Not inherited - occurs sporadically.
Pathognomonic appearance on X-ray: homogeneous ground-glass opacity with loss of normal trabecular pattern. Endosteal scalloping with cortical thinning (not breakthrough). No periosteal reaction unless fracture. Expansile lesion with hazy, smoky density.
Classic triad: polyostotic fibrous dysplasia, café-au-lait macules (coast of Maine irregular borders), and precocious puberty or other endocrinopathies (hyperthyroidism, Cushing, acromegaly). All from same GNAS mutation affecting different tissues.
Proximal femur varus deformity from repetitive microfractures and abnormal stress remodeling in polyostotic disease. Progressive coxa vara with lateral bowing. Prophylactic intramedullary fixation indicated to prevent progression and pathological fracture.
- Monostotic (70%)
- 70% of all cases
- Polyostotic (30%)
- 30% of cases
- Clinical Significance
- Monostotic 20:1 ratio in some series
- Monostotic (70%)
- Teens to 20s (adolescence)
- Polyostotic (30%)
- Childhood (under 10 years)
- Clinical Significance
- Polyostotic presents earlier
- Monostotic (70%)
- Ribs, femur, tibia, skull
- Polyostotic (30%)
- Craniofacial, femur, pelvis
- Clinical Significance
- Craniofacial involvement suggests polyostotic
- Monostotic (70%)
- None (isolated)
- Polyostotic (30%)
- McCune-Albright, Mazabraud
- Clinical Significance
- Polyostotic can have extraskeletal features
- Monostotic (70%)
- Stabilizes at skeletal maturity
- Polyostotic (30%)
- May progress through adulthood
- Clinical Significance
- Polyostotic requires lifelong surveillance
- Monostotic (70%)
- Observation or curettage/graft
- Polyostotic (30%)
- Bisphosphonates, prophylactic fixation
- Clinical Significance
- Polyostotic needs medical and surgical
Overview and Genetics
Fibrous dysplasia is a benign fibro-osseous developmental disorder characterized by replacement of normal medullary bone with fibrous tissue and immature woven bone. It is caused by a postzygotic somatic mutation in the GNAS gene, leading to abnormal bone remodeling and structural weakness.
Genetic Basis
- Postzygotic somatic mutation at Arg201 codon of GNAS gene
- Causes constitutive activation of Gsα protein
- Leads to excess cyclic AMP (cAMP) production
- Results in abnormal osteoblast differentiation
- Not inherited - occurs sporadically during early development
- Replacement of normal bone with fibrous tissue
- Production of immature woven bone (not lamellar)
- Loss of normal trabecular architecture
- Structural weakness and deformity
- Variable expressivity based on timing of mutation
Fibrous dysplasia is a developmental disorder, not a neoplasm, because it arises from a somatic mutation affecting bone development rather than uncontrolled cell proliferation. Lesions typically stabilize at skeletal maturity (monostotic form), supporting developmental rather than neoplastic nature.
Epidemiology
- Accounts for 5-7% of benign bone tumors
- No sex predilection for monostotic disease
- Slight female predominance for polyostotic (McCune-Albright)
- Monostotic: presents in adolescence to young adulthood (teens to 20s)
- Polyostotic: presents in childhood (often under 10 years)
- Lesions usually stabilize after skeletal maturity
- Monostotic (70%): single bone involvement
- Polyostotic (30%): multiple bones
- Craniofacial (25%): skull and facial bones common in polyostotic
Pathophysiology and Histology
Molecular Pathophysiology
- Mutation at Arg201: Prevents GTPase activity of Gsα
- Constitutive activation: Gsα remains in active GTP-bound state
- Excess cAMP: Adenylyl cyclase continuously activated
- Abnormal signaling: Dysregulated osteoblast function
- Fibro-osseous metaplasia: Normal marrow replaced by fibrous tissue
- Increased RANKL: Promotes osteoclast activity
- Decreased OPG: Reduces inhibition of bone resorption
- Abnormal osteoblasts: Produce immature woven bone
- Structural weakness: Loss of normal lamellar architecture
Gross Pathology
Macroscopic appearance:
- White-tan fibrous tissue replacing marrow
- Gritty texture from bone spicules
- No distinct capsule - blends with normal bone
- Vascular stroma - can be hemorrhagic
Histology
- Irregular bone trabeculae: C-shaped, S-shaped, alphabet-like
- Woven bone: Immature bone, not lamellar
- No osteoblastic rimming: Key differentiator from ossifying fibroma
- Fibrous stroma: Bland spindle cells, fibroblastic
- Variable bone content: From minimal to extensive
- Ossifying fibroma: Has osteoblastic rimming on trabeculae
- Low-grade osteosarcoma: Cytologic atypia, permeative growth
- Osteofibrous dysplasia: Cortical location, osteoblastic rimming
The Chinese letters pattern (irregular woven bone trabeculae) WITHOUT osteoblastic rimming is pathognomonic for fibrous dysplasia. Ossifying fibroma has similar trabeculae but WITH prominent osteoblastic rimming.
Clinical Forms and Syndromes
Monostotic Fibrous Dysplasia
Single bone involvement (70% of cases)
- Ribs (most common monostotic site)
- Proximal femur
- Tibia
- Craniofacial bones
- Humerus
- Often asymptomatic (incidental finding)
- Pathological fracture through weakened bone
- Bone pain (dull, aching)
- Deformity if progressive (rare)
- Stabilizes at skeletal maturity in most cases
Polyostotic Fibrous Dysplasia
Multiple bone involvement (30% of cases)
- Unilateral predominance (especially in McCune-Albright)
- Craniofacial involvement common
- Long bones (femur, tibia) frequently affected
- May involve 20-30+ bones in severe cases
- Earlier onset than monostotic (childhood)
- Progressive deformity (shepherd's crook)
- Multiple fractures
- Limb length discrepancy
- Cranial nerve compression if craniofacial
McCune-Albright Syndrome
- Polyostotic fibrous dysplasia (unilateral predominance)
- Café-au-lait macules (irregular "coast of Maine" borders)
- Endocrinopathy (precocious puberty most common)
- Precocious puberty (most common, especially in girls; recurrent ovarian cysts)
- Hyperthyroidism (autonomous nodular thyroid disease)
- Growth hormone excess / acromegaly (worsens craniofacial disease and optic compromise)
- Cushing syndrome (neonatal, due to adrenal hyperactivity)
- FGF23-mediated hypophosphataemia — excess FGF23 from dysplastic bone causes renal phosphate wasting; correlates with skeletal disease burden, worsens osteomalacia and fracture risk, and is an important treatable contributor to bone pain
Dysplastic FD bone over-produces FGF23, driving renal phosphate wasting and hypophosphataemic osteomalacia. This compounds bone weakness and fracture risk. Always check serum phosphate in extensive/polyostotic disease and replace phosphate and active vitamin D when low — an easily missed, treatable factor.
Dermatologic features:
- Café-au-lait spots with irregular borders
- Coast of Maine appearance (vs smooth "coast of California" in neurofibromatosis)
- Unilateral distribution following Blaschko lines
- Present at birth or early infancy
McCune-Albright syndrome requires multidisciplinary management involving orthopaedics, endocrinology, and genetics. Screen for endocrinopathies with thyroid function, cortisol, growth hormone, and bone age studies.
Mazabraud Syndrome
Fibrous dysplasia (monostotic or polyostotic) associated with intramuscular myxomas
- Intramuscular myxomas in skeletal muscle
- Same GNAS mutation as fibrous dysplasia
- Soft tissue masses adjacent to affected bones
- Rare syndrome (fewer than 100 reported cases)
Clinical Presentation and Examination
Presenting Symptoms
- Asymptomatic (50-70% of cases, incidental finding)
- Bone pain (dull, aching, worse with activity)
- Pathological fracture (sudden pain, deformity)
- Swelling if expansile (craniofacial lesions)
- Progressive deformity (shepherd's crook, limb bowing)
- Multiple fractures (recurrent, different sites)
- Limb length discrepancy (unilateral involvement)
- Gait abnormality (limp, antalgic gait)
- Cranial nerve symptoms (vision, hearing loss)
Physical Examination
- Visible deformity (varus femur, tibial bowing)
- Limb length discrepancy (measure true vs apparent length)
- Café-au-lait macules (check for McCune-Albright)
- Gait assessment (antalgic, Trendelenburg)
- Bony expansion over affected area
- Tenderness if symptomatic
- Warmth not typical (unless fracture)
- Shepherd's crook deformity: Proximal femur varus with lateral bowing
- Leonine facies: Craniofacial expansion in severe polyostotic
- Limb atrophy: If chronic pain/disuse
Investigations and Imaging
Radiography
- Homogeneous hazy density - pathognomonic finding
- Loss of normal trabecular pattern
- Smoky, frosted glass appearance
- Well-defined margins
- Endosteal scalloping with cortical thinning
- Expansion of medullary cavity
- Loss of corticomedullary differentiation
- No periosteal reaction (unless fracture)
- Shepherd's crook deformity - varus with lateral bowing
- Involves metaphysis and diaphysis
- Pathological fractures common
- Expansion of diploe
- Loss of normal contour
- Can cause orbital/sinus obliteration
- Expansile lucency with ground-glass matrix
- Most common monostotic site
CT Scan
- Evaluate extent of craniofacial involvement
- Assess cortical thickness for fracture risk
- Pre-operative planning for curettage
- Ground-glass attenuation (100-150 HU)
- Endosteal scalloping clearly visible
- 3D reconstruction helpful for deformity assessment
MRI
- Differentiate from malignancy if atypical
- Assess soft tissue extension (Mazabraud syndrome)
- Evaluate spinal canal involvement
- T1: Low to intermediate signal
- T2: Variable (low to high depending on fibrous vs cystic content)
- STIR: High signal (edema-like)
- Enhancement: Mild to moderate, heterogeneous
Bone Scan (Tc-99m)
Utility:
- Screen for polyostotic disease when single lesion found
- Shows increased uptake in active lesions
- Helps identify all sites of involvement
Laboratory Tests
Serum markers:
- Alkaline phosphatase: Often elevated (reflects bone turnover)
- Calcium: Usually normal
- Phosphate: May be low from FGF23-mediated renal phosphate wasting in extensive disease — check actively
- Endocrine screening if McCune-Albright suspected:
- Thyroid function (TSH, T4)
- Cortisol (24-hour urine free cortisol)
- Growth hormone, IGF-1
- Bone age (if precocious puberty)
Biopsy
- Atypical radiographic features (rule out malignancy)
- Pain without fracture (concern for sarcomatous transformation)
- Progressive lesion after skeletal maturity
- First diagnosis in polyostotic disease
- Core needle biopsy usually sufficient
- Open biopsy if inadequate sample
- Send for histology (Chinese letters pattern)

Differential Diagnosis
- Fibrous Dysplasia
- Any bone, ribs/femur common
- Ossifying Fibroma
- Mandible/maxilla only
- Osteofibrous Dysplasia
- Tibia/fibula cortex
- Fibrous Dysplasia
- Chinese letters, NO rimming
- Ossifying Fibroma
- Chinese letters WITH rimming
- Osteofibrous Dysplasia
- Cortical lesion, osteoblastic rimming
- Fibrous Dysplasia
- Ground-glass, medullary
- Ossifying Fibroma
- Well-defined, mixed density
- Osteofibrous Dysplasia
- Intracortical lucency, sclerotic rim
- Fibrous Dysplasia
- Teens to 20s (monostotic)
- Ossifying Fibroma
- 20-40 years
- Osteofibrous Dysplasia
- Under 10 years (children)
- Fibrous Dysplasia
- Stabilizes at maturity
- Ossifying Fibroma
- Slowly progressive
- Osteofibrous Dysplasia
- Can regress spontaneously
- Older age (over 50 years)
- Blade of grass advancing lytic front
- Cotton wool appearance in later stages
- Elevated alkaline phosphatase (markedly high)
- Eccentric location
- Fluid-fluid levels on MRI
- Blown-out appearance on X-ray
- No ground-glass matrix
- Permeative growth pattern
- Cytologic atypia on histology
- Periosteal reaction (Codman, sunburst)
- Older age or history of radiation
A craniofacial fibro-osseous lesion the differential above omits is cherubism, which an examiner may use to test that you do not reflexively attribute every fibro-osseous jaw lesion to fibrous dysplasia:
- Genetics: autosomal dominant, caused by SH3BP2 mutations - fundamentally different from the sporadic, somatic GNAS mutation of FD (so cherubism is inherited, FD is not).
- Presentation: bilateral, symmetric, painless expansion of the mandible and maxilla in a young child, classically giving full cheeks and the "eyes turned to heaven" (cherubic) appearance; FD is more often unilateral/asymmetric.
- Histology/imaging: multilocular giant-cell-rich fibrous lesions (not the woven-bone "Chinese letters" of FD), typically multicystic/lytic on imaging rather than ground-glass.
- Natural history: characteristically stabilises and regresses after puberty, so management is usually observation/conservative contouring with surgery reserved for functional or severe cosmetic problems - quite different from the progressive deformity of polyostotic FD.
Exam point: a bilateral symmetric jaw lesion in a child with a positive family history is cherubism (SH3BP2, autosomal dominant, giant-cell-rich, self-limiting after puberty) - not GNAS fibrous dysplasia, which is sporadic, often unilateral and shows ground-glass/Chinese-letters morphology.
Management
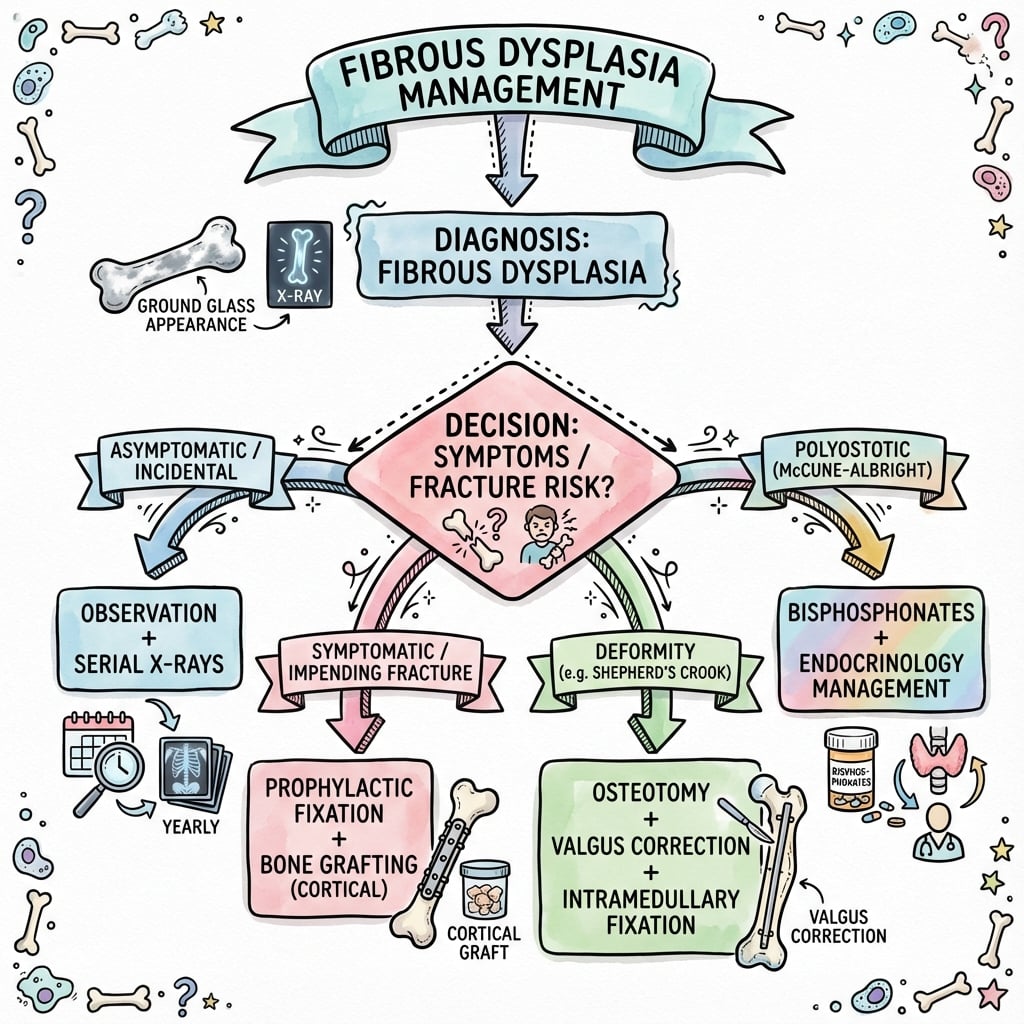
Conservative Management
- Asymptomatic monostotic lesions
- Small lesions without fracture risk
- Stable disease after skeletal maturity
- X-rays every 6-12 months initially
- Annual imaging once stable
- Monitor for growth, pain, deformity
Medical Therapy
- Symptomatic polyostotic disease
- Bone pain not responding to NSAIDs
- Reduce fracture risk in high-risk sites
- Pamidronate: 30-60 mg IV every 3-6 months
- Zoledronic acid: 4-5 mg IV annually
- Duration: Several years, reassess periodically
- Reduces bone pain in 60-80% of patients
- Decreases alkaline phosphatase levels
- May reduce fracture risk (limited data)
- Does not reverse deformity
- Flu-like symptoms after infusion
- Hypocalcemia (supplement calcium/vitamin D)
- Osteonecrosis of jaw (rare, dental hygiene important)
Bisphosphonates inhibit osteoclast activity and reduce bone turnover. Note the nuance: the placebo-controlled alendronate RCT (Boyce 2014) reduced resorption markers and raised bone density but did not significantly improve pain or function. International consensus therefore reserves them for refractory or fracture-related bone pain, not as disease modification.
Recent advances / controversies:
- Denosumab (RANKL inhibitor) has shown lesion regression in case reports of aggressive/refractory FD but has no controlled evidence and carries a serious rebound-hypercalcaemia risk on discontinuation, particularly in children — use only in specialist hands.
- Tocilizumab (anti-IL-6) is under investigation for FD-related pain.
- Phosphate and active vitamin D replacement for FGF23-mediated hypophosphataemia is an under-used, evidence-based adjunct that addresses a treatable cause of pain and osteomalacia.
Surgical Management
Indications for surgery:
- Pathological fracture (treat fracture, then address lesion)
- Impending fracture (cortical thinning over 50%, over 2.5cm lesion)
- Progressive deformity (shepherd's crook)
- Neurological compromise (craniofacial lesions)
- Refractory pain despite medical therapy
Curettage and Bone Grafting
- Symptomatic monostotic lesions
- After fracture healing
- Small, accessible lesions
- Exposure: Direct approach to lesion
- Curettage: Thorough removal of fibrous tissue
- Adjuvants: Phenol, argon beam, PMMA optional
- Grafting: Autograft or allograft cancellous bone
- Stability: Internal fixation if needed
- Recurrence rate: 10-30% in children, lower in adults
- Pain relief: 70-90% success rate
- Recurrence higher if lesion not fully mature
- Recurrence (especially in polyostotic)
- Fracture through graft site
- Infection (1-2%)
This completes the curettage technique section.
Treatment Algorithm
- Asymptomatic monostotic: Observe with serial X-rays
- Symptomatic monostotic: NSAIDs, consider bisphosphonates or surgery
- Polyostotic non-syndromic: Bisphosphonates, prophylactic fixation as needed
- McCune-Albright syndrome: Endocrine management, bisphosphonates, selective surgery
- Pathological fracture: Treat fracture conservatively, then address lesion after healing
- After skeletal maturity when possible (lower recurrence)
- Emergency: Fractures, neurological compromise
- Prophylactic: High-risk proximal femur lesions
Complications and Prognosis
Complications
Skeletal complications:
Pathological fracture (most common):
- Occurs in 20-50% of polyostotic patients
- Most common in proximal femur and tibia
- Usually minimally displaced
- Treat conservatively if possible (cast/brace)
- Consider prophylactic fixation after healing
- Shepherd's crook - proximal femur varus
- Limb length discrepancy - up to 5-10 cm in severe cases
- Angular deformity - tibial bowing
- Optic nerve - vision loss (10% of craniofacial cases)
- Auditory nerve - hearing loss
- Facial nerve - facial weakness
- Surgical decompression if progressive
The site list above (ribs, femur, skull, craniofacial) omits the spine, but vertebral involvement is well recognised in polyostotic disease and is an examinable, easily-missed complication:
- Scoliosis is the commonest spinal manifestation and a recognised cause of progressive deformity in polyostotic FD / McCune-Albright syndrome - curves can progress, contribute to disability, and (in severe untreated MAS) are associated with increased morbidity and even mortality, so active spinal surveillance is part of polyostotic-disease follow-up.
- Vertebral lesions (ground-glass, expansile) can cause pathological vertebral fracture and, rarely, spinal canal compromise/cord or root compression requiring decompression and stabilisation.
- Management: bracing has limited efficacy in dysplastic curves; posterior instrumented fusion is used for progressive or large curves, recognising the poorer bone quality and graft-resorption tendency of FD bone (favour instrumented load-sharing constructs, as in long bones).
Exam point: in polyostotic FD / McCune-Albright, examine and survey the spine - scoliosis is the principal spinal manifestation (and a contributor to morbidity), vertebral lesions can fracture or rarely compress neural elements, and surgical correction follows FD bone-quality principles rather than idiopathic-scoliosis assumptions.
Malignant Transformation
- Under 1% at population level (estimates commonly quoted as 0.4-1%)
- Referral series over-represent the risk: the Mayo Clinic cohort found 28 sarcomas among 1122 cases (Ruggieri 1994)
- Higher with prior radiotherapy — 46% of transformations in that series had received radiotherapy
- Slightly higher in polyostotic and craniofacial disease
- Radiation therapy (major avoidable risk factor)
- Polyostotic disease (slightly higher risk)
- Craniofacial lesions (commonest site of transformation alongside proximal femur)
- Osteosarcoma (most common, 19/28)
- Fibrosarcoma (5/28)
- Chondrosarcoma (3/28)
- Malignant fibrous histiocytoma (1/28)
- New pain in long-standing lesion
- Rapid growth after skeletal maturity
- Soft tissue mass on imaging
- Periosteal reaction or cortical destruction
Avoid radiation therapy for fibrous dysplasia: in the Mayo Clinic series of malignant transformation, 46% of patients who developed a sarcoma had received prior radiotherapy. Manage with surgery and, for refractory pain, bisphosphonates instead.
Prognosis
- Excellent prognosis - lesions stabilize at skeletal maturity
- Surgery curative in most cases
- Recurrence low after maturity (under 10%)
- Variable prognosis depending on extent
- May progress through adulthood
- Bisphosphonates improve quality of life
- Functional outcomes good with management
- Prognosis depends on endocrine complications
- Skeletal disease managed similarly to polyostotic
- Endocrinopathy management critical
- Multidisciplinary follow-up lifelong
Guidelines, Registries & Global Practice
Global Epidemiology
Fibrous dysplasia is rare, with an estimated prevalence in the region of 1 in 4,000 to 1 in 10,000, accounting for roughly 5-7% of benign bone tumours. McCune-Albright syndrome is far rarer (estimated between 1 in 100,000 and 1 in 1,000,000). The full FD/MAS spectrum arises from the same somatic gain-of-function GNAS mutation; monostotic disease predominates (~70-80% of cases) and is distributed worldwide without strong geographic or ethnic clustering. Because the disorder presents across orthopaedics, endocrinology, ENT/craniofacial surgery and dentistry, under-recognition and wide variation in investigation and treatment are well documented internationally — the central problem the 2019 international consensus set out to address.
Side-by-Side Guidance
- Scope / Focus
- Definitive multidisciplinary best-practice consensus
- Key positions
- GNAS-informed diagnosis, endocrine screening in MAS, surgery for deformity/fracture (not lesion eradication), bisphosphonates only for refractory bone pain
- Evidence level
- Consensus statement (expert)
- Scope / Focus
- Medical therapy and endocrinopathy management
- Key positions
- Treat MAS endocrinopathies (precocious puberty, GH excess, thyroid, FGF23-mediated hypophosphataemia); RCT data show bisphosphonates reduce turnover but not proven to relieve pain
- Evidence level
- RCT-informed (Level II for bisphosphonates)
- Scope / Focus
- Surgical strategy and oncological vigilance
- Key positions
- Load-sharing intramedullary fixation over plates in long bones; correct deformity by osteotomy; avoid radiotherapy; biopsy any lesion with red-flag change
- Evidence level
- Largely Level IV (case series)
- Scope / Focus
- Optic nerve and craniofacial disease
- Key positions
- Prophylactic optic-nerve decompression NOT routinely recommended for asymptomatic encasement; operate for documented progressive visual loss; conservative contouring over radical resection
- Evidence level
- Consensus (expert)
Multidisciplinary management: Complex FD, and McCune-Albright syndrome in particular, requires coordinated orthopaedic surgery, endocrinology, ENT/craniofacial surgery, ophthalmology and genetics input. The consensus model is a designated multidisciplinary team or rare-bone-disease referral centre, an approach mirrored across European reference networks (e.g. ERN BOND) and major North American and Australasian paediatric centres.
Bisphosphonate access and use: Intravenous pamidronate and zoledronic acid (and oral alendronate) are widely available; international consensus restricts their use to fracture-related or persistent bone pain rather than disease modification, reflecting the Boyce 2014 RCT. Denosumab has been used off-label in refractory disease but lacks controlled evidence and carries a notable rebound-hypercalcaemia risk, especially in children. Intravenous bisphosphonates are administered in a hospital or specialist-supervised setting; the funding and access route for infused agents varies between health systems, but the clinical indication — specialist-supervised use for refractory or fracture-related bone pain — is consistent internationally.
Genetic testing: Somatic GNAS mutation testing on lesional tissue (or, with sensitive assays, peripheral blood in MAS) can confirm atypical cases. It is used for diagnostic confirmation and counselling — the disorder is sporadic and not heritable, so cascade family testing is not indicated.
Surveillance and registries: Plain radiographs remain first-line; CT aids craniofacial/surgical planning and bone scintigraphy or whole-body MRI maps polyostotic burden. There is no large dedicated FD/MAS arthroplasty-style registry; longitudinal cohorts (notably the NIH natural-history cohort) and the FD/MAS consortium provide the principal pooled outcome data, and patient organisations maintain international registries to support rare-disease research.
MCQ Practice Points
Q: What mutation causes fibrous dysplasia and what is its mechanism?
A: GNAS mutation at Arg201 codon (postzygotic somatic mutation) causing constitutive activation of Gsα protein and excess cAMP production. This drives abnormal osteoblast differentiation, replacing normal bone with fibrous tissue and immature woven bone. It is NOT inherited - occurs sporadically.
Q: What is the pathognomonic radiographic appearance of fibrous dysplasia?
A: Ground-glass opacity with homogeneous, hazy, smoky density. Loss of normal trabecular pattern with endosteal scalloping and cortical thinning (not cortical breakthrough). The lesion is expansile but well-circumscribed. No periosteal reaction unless complicated by fracture.
Q: What are the three classic features of McCune-Albright syndrome?
A: Polyostotic fibrous dysplasia plus café-au-lait macules (coast of Maine borders, irregular) plus precocious puberty (or other endocrinopathy such as hyperthyroidism, acromegaly, Cushing syndrome). All result from the same GNAS mutation affecting multiple tissue types.
Q: What is the classic histological pattern of fibrous dysplasia?
A: Chinese letters (alphabet soup) pattern - irregular trabeculae of woven bone in a fibrous stroma WITHOUT osteoblastic rimming. Absence of osteoblastic rimming distinguishes from reactive bone or ossifying fibroma. Trabeculae curve and branch in irregular patterns resembling Chinese characters.
At a Glance
Fibrous dysplasia is a benign fibro-osseous developmental disorder (not a true neoplasm) caused by a postzygotic somatic GNAS mutation at Arg201 codon, leading to constitutive cAMP activation and replacement of normal bone with fibrous tissue and immature woven bone. Monostotic disease (70%) affects ribs/femur and stabilizes at skeletal maturity; polyostotic (30%) presents earlier in childhood and may progress. The ground-glass radiographic appearance with endosteal scalloping is pathognomonic. Shepherd's crook deformity (proximal femur varus) results from repetitive microfractures in polyostotic disease. McCune-Albright syndrome comprises polyostotic FD, café-au-lait macules (coast of Maine irregular borders), and precocious puberty. Histology shows Chinese letters pattern—irregular woven bone without osteoblastic rimming.
FIBROUSFibrous Dysplasia Key Features
Hook:Think FIBROUS tissue replacing bone - ground-glass appearance is classic!
CAFEMcCune-Albright Syndrome Triad
Hook:Drink CAFE and think of café-au-lait spots - McCune-Albright syndrome!
CHINESEHistology - Chinese Letters
Hook:CHINESE letters = irregular bone trabeculae that look like alphabet characters!
STOPTreatment Options - STOP
Hook:STOP and think - most monostotic lesions need observation only!
Exam Viva Scenarios
Practise clinical reasoning and management decisions out loud
“A 25-year-old female undergoes chest X-ray for pneumonia screening. An incidental well-defined expansile lesion with ground-glass appearance is noted in the left 6th rib. She is completely asymptomatic. How would you manage this patient?”
“A 12-year-old boy with known polyostotic fibrous dysplasia presents with progressive left proximal femur deformity and limp. X-rays show shepherd's crook deformity with varus angulation and ground-glass appearance involving the proximal third of the femur. How would you approach this case?”
“A 45-year-old man with known fibrous dysplasia of the proximal humerus for 20 years presents with new onset pain over the past 3 months without trauma. X-rays show the known ground-glass lesion but with some ill-defined margins and possible soft tissue fullness. What are your concerns and how would you proceed?”
Genetics & Pathophysiology
- **GNAS mutation** at Arg201 codon - postzygotic somatic mutation
- **Constitutive Gsα activation** → excess cAMP → abnormal osteoblast function
- **NOT inherited** - sporadic developmental disorder, not true neoplasm
- **Fibro-osseous metaplasia** - normal marrow replaced by fibrous tissue and woven bone
Clinical Forms (3 Types)
- **Monostotic (70%)**: single bone, ribs/femur, teens-20s, stabilizes at maturity
- **Polyostotic (30%)**: multiple bones, childhood onset, craniofacial common
- **McCune-Albright**: polyostotic FD + café-au-lait (coast of Maine) + endocrine (precocious puberty)
Key Imaging Features
- **Ground-glass opacity** - homogeneous hazy density (pathognomonic)
- **Endosteal scalloping** with cortical thinning (not breakthrough)
- **Loss of corticomedullary differentiation** - blurred inner cortex
- **No periosteal reaction** unless fracture present
- **Shepherd's crook deformity** - proximal femur varus in polyostotic
Histology - Chinese Letters
- **Irregular woven bone trabeculae** - C, S, alphabet shapes
- **NO osteoblastic rimming** - key vs ossifying fibroma (has rimming)
- **Fibrous stroma** - bland spindle cells, fibroblastic background
- **Variable bone content** - minimal to extensive woven bone
Management Principles
- **Asymptomatic monostotic**: observe with serial X-rays (most common)
- **Bisphosphonates** (pamidronate, zoledronic acid): for pain in polyostotic disease
- **Curettage + bone graft**: symptomatic monostotic after fracture
- **Prophylactic IM nail**: shepherd's crook, impending fracture (over 50% cortical)
- **Avoid radiation therapy**: major avoidable risk for malignant transformation
Complications & Prognosis
- **Pathological fracture** - most common complication (20-50% polyostotic)
- **Malignant transformation** - under 0.5% overall, osteosarcoma most common
- **Cranial nerve compression** - vision/hearing loss in craniofacial (10%)
- **Monostotic prognosis excellent** - stabilizes at skeletal maturity
- **Polyostotic variable** - may progress, bisphosphonates improve QOL
Exam Viva Key Points
- **Developmental disorder NOT neoplasm** - postzygotic mutation affects development
- **Ground-glass = pathognomonic** - differentiates from most other lesions
- **Monostotic 20:1** more common than polyostotic
- **Ossifying fibroma has osteoblastic rimming**, FD does not
- **McCune-Albright triad**: FD + café-au-lait + endocrine (CAFE mnemonic)
Evidence Base
Activating Mutations of the Stimulatory G Protein in McCune-Albright Syndrome
- Activating Gsα (GNAS) mutations at codon Arg201 (R201H and R201C) identified in all 4 MAS patients
- Mosaic tissue distribution supports a postzygotic somatic mutation early in embryogenesis
- Loss of GTPase activity drives constitutive Gsα activation and excess cAMP
- Established the unifying genetic basis for fibrous dysplasia and McCune-Albright syndrome
GNAS: Normal and Abnormal Functions (Gsα signalling and imprinting)
- Constitutive Gsα activation results from impaired GTP hydrolysis at Arg201/Gln227
- Same gene produces opposite phenotypes: activating → FD/MAS, inactivating → AHO
- Tissue-specific GNAS imprinting helps explain the variable endocrine phenotype
- Provides the molecular framework linking cAMP excess to abnormal osteoblast differentiation
Randomised Controlled Trial of Alendronate for Fibrous Dysplasia of Bone
- Alendronate lowered urinary NTX-telopeptide and raised areal BMD in FD lesions (both statistically significant)
- No significant effect on pain scores, skeletal disease burden or functional walk/strength tests
- Serum osteocalcin unchanged between groups
- Best evidence that bisphosphonates modulate bone turnover but are not proven to relieve FD pain or alter the lesion radiographically
Best Practice Management Guidelines for Fibrous Dysplasia / McCune-Albright Syndrome
- Defines best-practice diagnostic and monitoring pathways across orthopaedics, endocrinology and craniofacial care
- Surgery aims to correct/prevent deformity and fracture, not to 'cure' the lesion (grafts characteristically resorb)
- Bisphosphonates recommended for fracture-related or persistent bone pain, not as disease-modifying therapy
- Mandates endocrine screening and prophylactic optic-nerve management decisions in craniofacial MAS
Sarcomatous (Malignant) Transformation in Fibrous Dysplasia
- 28 sarcomas in 1122 referral cases of FD; osteosarcoma the most common transformation (19/28)
- 46% of transformations followed prior radiotherapy — a key avoidable risk factor
- Craniofacial bones and proximal femur were the commonest sites of malignant change
- Population-level malignant transformation risk is under 1%; this referral cohort over-represents it
Curettage, Bone-Grafting and Realignment for Proximal Femoral Fibrous Dysplasia
- 22 patients / 27 femora; grafts uniformly resorbed and lesions persisted
- Curettage + grafting offered no advantage over realignment osteotomy alone
- Valgus osteotomy with internal fixation, performed early, is the treatment of choice for shepherd's crook deformity
- Polyostotic disease with calcar involvement had worse bone quality and outcomes