Charcot-Marie-Tooth Disease | CMT
- Definition: Group of inherited peripheral neuropathies. Most common inherited neuropathy.
- Genetics: CMT1A (PMP22 duplication) is most common (50%). Autosomal dominant.
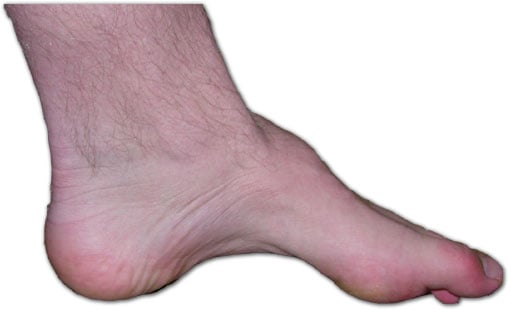
- Foot Deformity: Cavovarus foot - High arch, Hindfoot varus, Claw toes.
- Coleman Block Test: Differentiates FIXED from FLEXIBLE hindfoot varus.
- Management: Non-op (Orthotics, PT) then Osteotomies if progressive/fixed.
- “CMT is the most common inherited neuropathy.
- “Cavovarus foot = Weak Peroneus Brevis (Tibialis Posterior unopposed).
- “Coleman Block Test: If hindfoot corrects when 1st ray offloaded, varus is forefoot-driven.
- “Surgery: Soft tissue balancing + Osteotomies (Calcaneal, Midfoot, 1st MT).
Think CMT with Cavovarus. Any patient with cavovarus foot and weakness should be evaluated for CMT.
Critical Test. Determines if surgery should address forefoot (1st MT) or hindfoot (Calcaneal osteotomy).
Expect Progression. Surgery may need revision. Long-term follow-up essential.
Check the Spine. Scoliosis occurs in CMT. Screen and monitor.
- Pathology
- Demyelinating
- NCS
- Slow velocity
- Genetics
- PMP22 Duplication (AD)
- Pathology
- Demyelinating
- NCS
- Slow velocity
- Genetics
- MPZ mutation (AD)
- Pathology
- Axonal
- NCS
- Normal velocity, Low amplitude
- Genetics
- MFN2 mutation (AD)
- Pathology
- Axonal
- NCS
- Variable
- Genetics
- Connexin 32 (X-linked)
CHARCOTCMT Features
Hook:Key features of CMT.
BLOCKColeman Block Test
Hook:Coleman Block = Forefoot vs Hindfoot varus.
PB vs PTMuscle Imbalance
Hook:PB weakness causes varus.
Overview and Epidemiology
Hereditary Motor Sensory Neuropathies (HMSN), also known as Charcot-Marie-Tooth (CMT) disease, are a group of inherited peripheral neuropathies characterized by progressive distal muscle weakness and sensory loss.
- Incidence: 1 in 2500 (Most common inherited neuropathy).
- CMT1A: 50% of all CMT cases.
- Inheritance: Mostly Autosomal Dominant.
Named after Jean-Martin Charcot, Pierre Marie (France), and Howard Henry Tooth (UK) who independently described the condition in 1886.
Genetics
Genetic Basis of HMSN/CMT:
- Chromosome
- 17p11.2
- CMT Type
- CMT1A (50%)
- Inheritance
- AD
- Mechanism
- Demyelinating
- Chromosome
- 1q22
- CMT Type
- CMT1B
- Inheritance
- AD
- Mechanism
- Demyelinating
- Chromosome
- 1p36
- CMT Type
- CMT2A
- Inheritance
- AD
- Mechanism
- Axonal
- Chromosome
- Xq13
- CMT Type
- CMTX
- Inheritance
- X-linked
- Mechanism
- Mixed
- CMT1A (50%): PMP22 gene duplication (Chromosome 17). Most common cause.
- CMT1B: MPZ (Myelin Protein Zero) mutation. Demyelinating.
- CMT2A: MFN2 (Mitofusin 2) mutation. Axonal transport defect.
- CMTX: Connexin 32 mutation. X-linked inheritance.
- First-line: PMP22 duplication/deletion analysis (detects 70% of CMT1).
- Second-line: Targeted gene panels or whole exome sequencing.
- Prenatal testing available for known familial mutations.
The Allelic Counterpart: HNPP
The PMP22 gene that is duplicated in CMT1A is deleted in the reciprocal disorder hereditary neuropathy with liability to pressure palsies (HNPP) - a gene-dosage mirror image that examiners like to pair with CMT1A.
- Genetics: autosomal dominant deletion of one PMP22 allele on 17p11.2 (the same nonallelic homologous recombination event that, in the opposite direction, duplicates PMP22 to cause CMT1A).
- Clinical picture: recurrent, painless, often transient focal palsies precipitated by minor compression or stretch at common entrapment sites (the peroneal nerve at the fibular neck, ulnar at the elbow, median at the wrist, brachial plexus) - quite different from the slowly progressive symmetric cavovarus of CMT.
- Biopsy: focal sausage-shaped myelin thickenings called tomacula ("tomaculous neuropathy").
Q: A PMP22 duplication causes CMT1A - what does a PMP22 deletion cause? A: Hereditary neuropathy with liability to pressure palsies (HNPP), the reciprocal gene-dosage disorder. It presents with recurrent painless pressure palsies (for example a foot drop from peroneal compression at the fibular neck) rather than progressive cavovarus, and shows tomacula (sausage-like myelin) on nerve biopsy.
Anatomy and Biomechanics
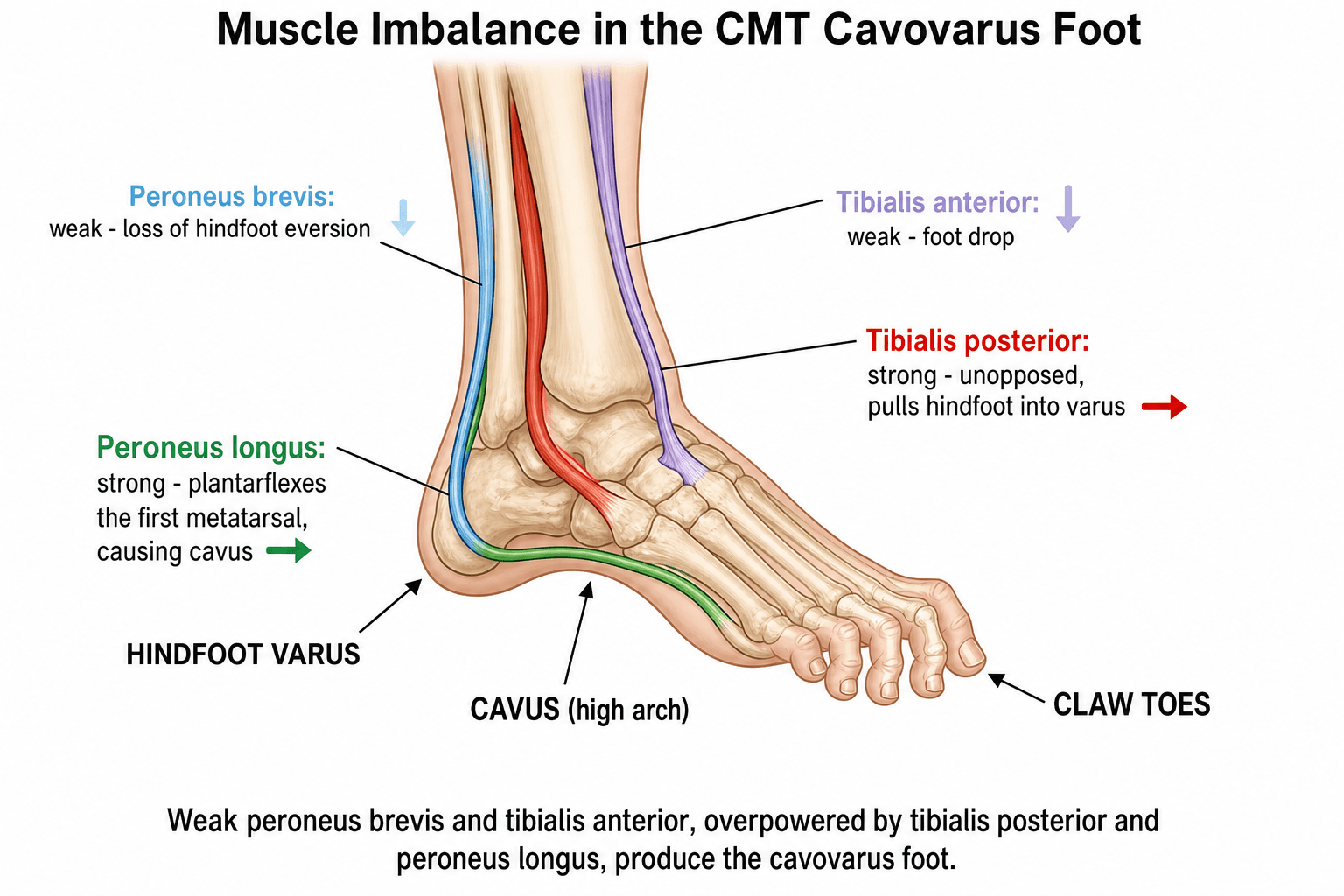
The "Tripod" Concept of the Cavovarus Foot: The forefoot is described as a tripod resting on the 1st metatarsal head, the 5th metatarsal head, and the heel. In CMT the strong peroneus longus plantarflexes the 1st metatarsal (1st ray), driving the medial leg of the tripod down. To keep all three points on the ground, the hindfoot compensates by rolling into varus. This is the mechanical basis of the "forefoot-driven" hindfoot varus that the Coleman block test unmasks.
Sequence of Muscle Imbalance (drives the deformity):
- Status in CMT
- Weak (early)
- Antagonist
- Tibialis posterior
- Deforming effect
- Loss of hindfoot eversion to varus
- Status in CMT
- Weak (later)
- Antagonist
- Peroneus longus
- Deforming effect
- Foot drop to forefoot equinus
- Status in CMT
- Relatively strong
- Antagonist
- Peroneus brevis
- Deforming effect
- Hindfoot inversion (varus)
- Status in CMT
- Relatively strong
- Antagonist
- Tibialis anterior
- Deforming effect
- 1st ray plantarflexion (cavus)
- Status in CMT
- Weak (early)
- Antagonist
- Long extensors/flexors
- Deforming effect
- Claw toes, loss of arch support
- Plantarflexed 1st ray to forefoot pronation and cavus.
- Forefoot-driven hindfoot varus (tripod compensation).
- Over time the hindfoot varus becomes fixed (subtalar contracture, calcaneal varus tilt).
- Varus heel laterally overloads the foot to recurrent ankle sprains, peroneal tendinopathy, 5th metatarsal stress fractures, and a varus ankle that can progress to medial ankle arthritis.
Correction must be staged from distal deforming force outward — release tight static structures (plantar fascia), neutralise the deforming dynamic force (peroneus longus to brevis transfer), correct the bony apex (1st metatarsal dorsiflexion osteotomy), and only then add a calcaneal osteotomy if the hindfoot remains varus once the forefoot is balanced.
Pathophysiology
Cellular and Molecular Mechanisms:
CMT pathophysiology depends on the specific genetic defect:
- Schwann Cell Dysfunction: PMP22 or MPZ mutations disrupt myelin formation.
- Demyelination/Remyelination Cycles: Repeated attempts at remyelination create characteristic 'onion bulb' formation on nerve biopsy.
- Result: Slowed nerve conduction velocities (less than 38 m/s).
- Clinical Effect: Progressive distal weakness and sensory loss.
- Axonal Degeneration: MFN2 mutations affect mitochondrial function and axonal transport.
- Wallerian Degeneration: Distal axon breakdown without primary demyelination.
- Result: Normal conduction velocities but reduced amplitude.
- Clinical Effect: Similar weakness pattern, often later onset.
The characteristic foot deformity results from selective muscle weakness:
- Peroneus Brevis: Weakens first (primary evertor of hindfoot).
- Tibialis Posterior: Relatively preserved. Unopposed inversion pulls hindfoot into varus.
- Peroneus Longus: Relatively preserved. Plantarflexes 1st metatarsal creating forefoot equinus.
- Intrinsic Muscles: Early weakness. Long flexors/extensors become dominant causing claw toes.
- Plantar Fascia: Secondary contracture contributes to arch elevation.
Net Result: Cavovarus foot (High arch + Hindfoot Varus + Claw toes + Forefoot adduction).
Progression Pattern:
- Distal-to-proximal weakness ('dying back' neuropathy).
- Lower limbs affected before upper limbs.
- Sensory loss follows similar pattern.
- Deformity progresses throughout growth and into adulthood.
Classification Systems
Dyck & Lambert Classification (electrophysiological)
The original HMSN classification is based on nerve conduction velocity (NCV), measured in the upper limb (median/ulnar motor):
- CMT1 / HMSN I (Demyelinating): Motor NCV under 38 m/s. PMP22 duplication most common. Onion bulbs on biopsy. Most common form (about 60% of cases).
- CMT2 / HMSN II (Axonal): Motor NCV over 38 m/s but reduced amplitude (axonal loss). Often later onset, more pure motor.
- Intermediate CMT: NCV 25 to 45 m/s (e.g. CMTX in males, DI-CMT).
- CMTX: X-linked, GJB1/connexin 32. Males more severely affected than females; intermediate NCV.
- CMT4 / HMSN III (Dejerine-Sottas): Autosomal recessive, severe, infantile onset, very slow NCV. Scoliosis is a hallmark (e.g. CMT4C / SH3TC2).
Clinical Assessment
- Family History: Autosomal dominant (often affected parent).
- Onset: Usually childhood/adolescence.
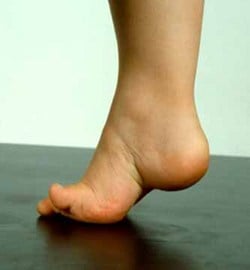
- Symptoms: Difficulty walking, frequent ankle sprains, foot drop, clumsiness.
- Inspection:
- 'Stork legs' (Distal wasting, Normal proximal).
- Cavovarus foot (High arch, Hindfoot varus).
- Claw toes.
- Calluses (Metatarsal heads, Lateral foot).
- Motor:
- Weak ankle dorsiflexion (Foot drop).
- Weak eversion (Peroneus Brevis).
- Weak toe extension/flexion (Intrinsics).
- Sensory: Glove-stocking sensory loss.
- Reflexes: Reduced/absent ankle jerks.
- Gait: Steppage gait (High stepping to clear foot).
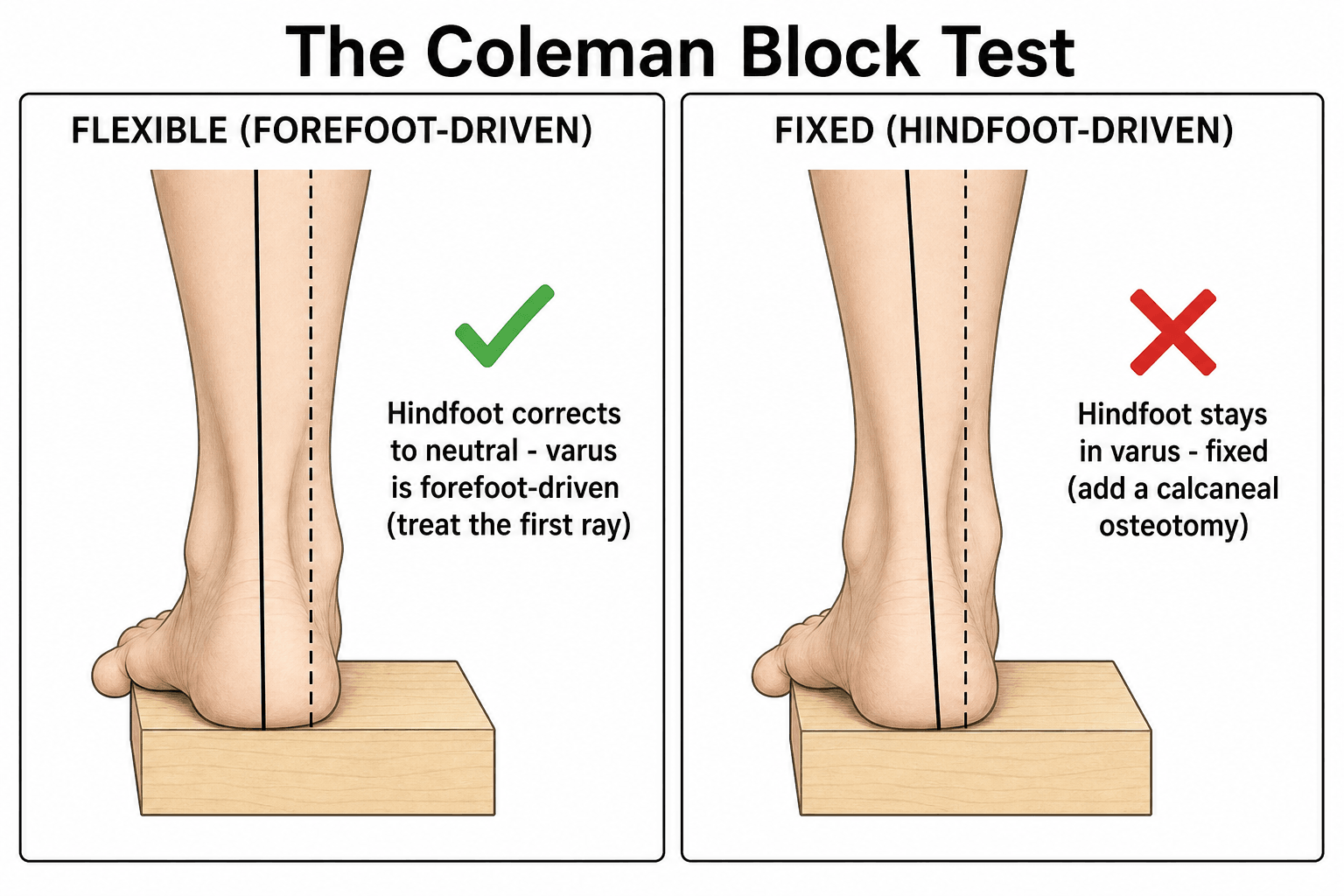
- Patient stands on a 2-3cm block.
- Lateral foot on block. 1st ray (1st MT head) hangs off medially.
- Observe Hindfoot: Does varus correct?
- If Corrects: Varus is FOREFOOT-DRIVEN (1st ray plantarflexion). Surgery addresses 1st ray.
- If Does Not Correct: Varus is FIXED/HINDFOOT. Surgery needs calcaneal osteotomy.
Investigations
- Clinical Examination: Often sufficient.
- NCS/EMG:
- CMT1: Slow conduction velocities (less than 38 m/s).
- CMT2: Normal velocity, Reduced amplitude.
- Genetic Testing: Confirms specific mutation (CMT1A = PMP22 duplication).
- Nerve Biopsy: Rarely needed. 'Onion bulbs' in CMT1.
- Weight-Bearing X-rays: AP, Lateral foot. Assess arch height, Meary's angle.
- Coleman Block Test: As above.
- Spine X-ray: Screen for scoliosis.
Beyond the Foot: Hip and Hand
CMT is a generalised neuropathy, so the foot is not the whole story - two examinable sites are easily overlooked.
- Hip dysplasia: acetabular dysplasia is a recognised association of CMT (reported in roughly the high single figures of patients) and is frequently missed because it is often painless. The threshold for a screening pelvis radiograph should be low, particularly in the growing child, because untreated dysplasia progresses to subluxation and early arthritis and is far easier to address when detected early (a peri-acetabular or femoral osteotomy) than late.
- The CMT hand: as the neuropathy ascends, the hand develops intrinsic-muscle wasting (first dorsal interosseous and thenar), a claw posture and weak pinch/grip, impairing fine tasks such as buttons and keys. Management is largely supportive (therapy, splints), with selective tendon transfers (for example to restore thumb opposition or correct clawing) in carefully chosen patients.
- Spine: scoliosis (already covered) is more common in early-onset and autosomal-recessive forms and warrants surveillance.
Q: Besides the foot and spine, what must you screen for in a child with CMT, and why is it easily missed? A: Acetabular (hip) dysplasia - a recognised CMT association that is often painless and therefore missed. A screening pelvis radiograph is warranted because early detection allows joint-preserving osteotomy, whereas late dysplasia progresses to subluxation and early arthritis. The hands also develop intrinsic wasting and clawing as the neuropathy ascends.
Differential Diagnosis
The cavovarus foot is the orthopaedic presentation, but a cavus foot is a red flag for an underlying neurological diagnosis until proven otherwise. Bilateral, symmetric, slowly progressive cavovarus with a positive family history points strongly to CMT, but the following must be considered and excluded.
- Distinguishing features
- Bilateral, symmetric, family history, distal wasting, areflexia, glove-stocking sensory loss
- Key investigation
- NCS/EMG + genetic testing (PMP22)
- Distinguishing features
- Ataxia, dysarthria, cardiomyopathy, diabetes, upgoing plantars (UMN signs)
- Key investigation
- FXN gene (GAA triplet repeat)
- Distinguishing features
- UNILATERAL cavus, back/midline skin signs, bladder symptoms, rapid progression
- Key investigation
- Whole-spine MRI
- Distinguishing features
- Spasticity, UMN signs, perinatal history, brisk reflexes
- Key investigation
- Clinical + brain MRI
- Distinguishing features
- Asymmetric, flail segments, no sensory loss, static (non-progressive)
- Key investigation
- History + EMG
- Distinguishing features
- Mild, non-progressive, normal neuro exam
- Key investigation
- Diagnosis of exclusion
A unilateral or rapidly progressive cavovarus foot is NOT typical CMT (which is bilateral and symmetric). It mandates whole-spine MRI to exclude tethered cord, diastematomyelia, syrinx, or an intraspinal tumour before any foot surgery.
Management Algorithm
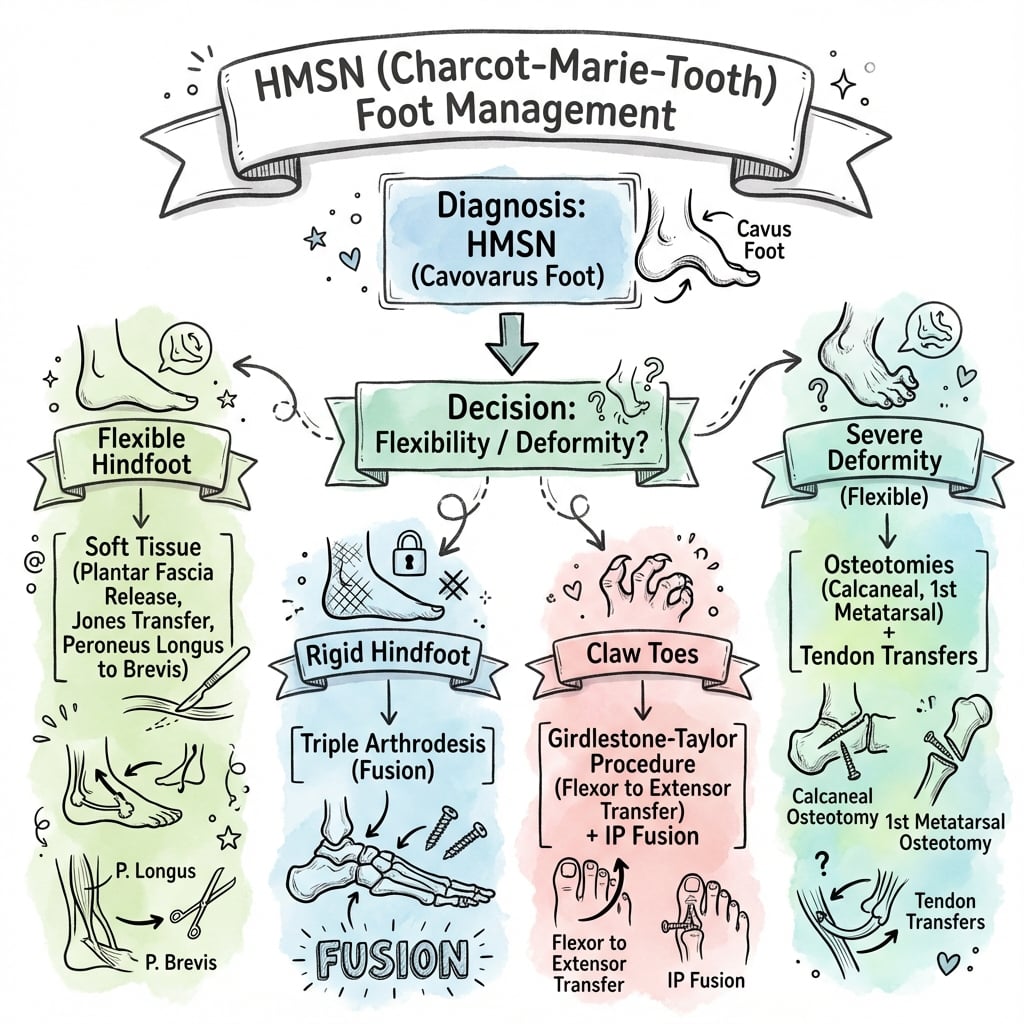
Non-Operative (Initial)
- Physiotherapy: Stretching (Plantar fascia, Achilles). Strengthening.
- Orthotics:
- AFO (Foot drop).
- Lateral wedge (Mild varus).
- Custom-moulded insoles.
- Activity Modification: Supportive footwear. Avoid high heels.
- Surveillance: Regular follow-up. Progression is expected.
Non-op suitable for mild, flexible deformity.
Surgical Technique
Lateral Calcaneal Osteotomy (Dwyer)
For fixed hindfoot varus.
- Incision: Lateral oblique over calcaneus.
- Protect: Sural nerve, Peroneal tendons.
- Osteotomy: Lateral closing wedge (remove wedge, base lateral).
- Fixation: Staple or Screw.
- Result: Hindfoot is shifted into neutral/slight valgus.
Often combined with lateralizing component (shift calcaneus laterally).
Complications
- Risk Factor
- Progressive disease
- Management
- Revision surgery
- Risk Factor
- Excessive osteotomy
- Management
- Revision / Accept
- Risk Factor
- Poor technique
- Management
- Revision fixation
- Risk Factor
- Fusion procedures
- Management
- Expected tradeoff
- Risk Factor
- Calcaneal osteotomy
- Management
- Careful dissection
Postoperative Care
- NWB Cast 6-8 weeks.
- Transition to AFO or supportive footwear.
- Physiotherapy for ROM and strengthening.
- Splint in corrected position 4-6 weeks.
- Gradual retraining of transferred muscles.
Outcomes
- Short-term: Reliable deformity correction is achievable with the combined soft-tissue + osteotomy ("joint-sparing") approach.
- Long-term: In the longest published series (mean 26-year follow-up of joint-sparing reconstruction), the cavus correction was well maintained but most feet showed some radiographic recurrence of hindfoot varus, reflecting the progressive nature of CMT. Despite this, no patient required salvage triple arthrodesis and rates of degenerative change and reoperation were lower than historically reported after triple arthrodesis.
- Function: Surgery improves plantigrade stance, reduces lateral-border overload and recurrent sprains, and improves gait efficiency. Patient-reported physical function (SF-36) remains below age-matched norms because of the underlying neuropathy.
- Smoking: An independent predictor of worse pain and disability scores after CMT cavovarus reconstruction — counsel cessation.
- Counselling: Patients (and parents) must understand that surgery corrects the deformity but does not cure the neuropathy; progression and possible revision should be anticipated.
Guidelines, Registries & Global Practice
Global epidemiology:
- CMT/HMSN is the most common inherited neuropathy, with a widely cited population prevalence of approximately 1 in 2,500 (around 1 in 1,200 to 1 in 3,300 across studies).
- CMT1A (PMP22 duplication) is the single most common subtype; in population-based genetic series the PMP22 duplication accounts for roughly 10 to 60% of genetically defined cases.
- Cavovarus foot deformity is the dominant orthopaedic manifestation; scoliosis is more frequent in early-onset and autosomal-recessive forms (e.g. CMT4C).
Side-by-side practice guidance (no single national framework governs CMT foot surgery):
- Emphasis
- Genetic confirmation, multidisciplinary care, CMTNS for monitoring; PMP22 duplication tested first
- Emphasis
- Joint-sparing osteotomy + tendon transfer for flexible deformity; arthrodesis reserved for rigid/arthritic feet
- Emphasis
- Stable internal fixation of osteotomies, plantigrade balanced foot, address all deformity components in one stage
- Emphasis
- Early soft-tissue balancing to prevent fixed bony deformity; spine surveillance for scoliosis
- No dedicated arthroplasty-style registry exists for CMT foot surgery; evidence is from specialist case series (e.g. the 26-year joint-sparing cohort).
- International CMT patient registries and natural-history consortia (e.g. the Inherited Neuropathies Consortium) underpin genotype-phenotype data and trial recruitment.
- High-resource settings: broad NGS gene panels, gait analysis, custom orthotics/AFOs, staged joint-sparing reconstruction, MDT neuromuscular clinics.
- Limited-resource settings: diagnosis often clinical plus basic nerve conduction studies; management weighted toward bracing and a single definitive corrective procedure; emerging genetic and disease-modifying therapies are largely inaccessible.
Controversies and Areas of Uncertainty
The pendulum has swung decisively toward joint-sparing osteotomies + tendon transfers in flexible/correctable feet, reserving triple arthrodesis for rigid, arthritic, or salvage feet. Long-term data favour the joint-sparing approach, but the optimal threshold for fusion remains debated.
Whether to operate early (flexible deformity, simpler soft-tissue balancing) or wait until skeletal maturity (deformity stable, fewer revisions) is unresolved. Many advocate early balancing to prevent fixed bony deformity.
The value of "prophylactic" tibialis posterior or anterior transfer in a still-flexible foot to slow deformity is not established by high-level evidence.
No drug is yet licensed to alter CMT progression. Gene-targeted and small-molecule therapies (e.g. PMP22-lowering approaches) are in trials; this is a moving field and may change orthopaedic relevance in coming years.
MCQ Practice Points
Q: What is the most common type of CMT? A: CMT1A (50% of cases). Caused by PMP22 gene duplication. Autosomal dominant.
Q: If the hindfoot varus corrects on Coleman Block Test, what does this indicate? A: The varus is FOREFOOT-DRIVEN (plantarflexed 1st ray). Surgical correction should address the 1st ray (Dorsiflexion 1st MT Osteotomy).
Q: What muscle imbalance causes cavovarus in CMT? A: Weak Peroneus Brevis (first to go) with relatively strong Tibialis Posterior (pulls into varus) and Peroneus Longus (plantarflexes 1st ray).
Q: What NCS finding is seen in CMT1 (demyelinating)? A: Slow nerve conduction velocity (less than 38 m/s motor).
Q: What is the purpose of Peroneus Longus to Brevis transfer in CMT? A: 1) Removes the deforming force on the 1st ray (reduces plantarflexion). 2) Augments the weak Peroneus Brevis (improves eversion).
Exam Viva Scenarios
Practise clinical reasoning and management decisions out loud
“What is your diagnosis and approach?”
“Demonstrate and explain the test.”
“Outline your surgical plan.”
Key Features
- Most common inherited neuropathy
- CMT1A = PMP22 duplication
- Cavovarus foot
- Weak PB, Strong PT/PL
Coleman Block
- Block under lateral foot
- 1st ray offloaded
- If corrects = Forefoot-driven
- If not = Hindfoot-driven
Surgery
- PF Release
- PL to PB Transfer
- 1st MT Osteotomy (if Coleman+)
- Calcaneal Osteotomy (if Coleman-)
- Jones Procedure (claw toes)
Pitfalls
- Progressive disease
- Recurrence common
- Check spine for scoliosis
Evidence Base
Joint-Sparing Reconstruction — 26-Year Follow-up
- 25 patients (41 feet) with CMT cavovarus treated with 1st metatarsal dorsiflexion osteotomy, peroneus longus to brevis transfer, plantar fascia release and EHL transfer; mean follow-up 26.1 years.
- Cavus correction was well maintained but most feet had radiographic recurrence of hindfoot varus.
- No patient required triple arthrodesis; lower degenerative change and reoperation than historical triple-arthrodesis series.
- Smoking was associated with significantly worse Foot Function Index pain and disability scores.
Molecular Basis of CMT1A — PMP22 Duplication
- Identified a large DNA duplication on chromosome 17p completely linked to CMT1A.
- Demonstrated three alleles at a polymorphic locus and a novel 500 kb SacII fragment in affected individuals.
- Established CMT1A as a gene-dosage disorder from inherited DNA rearrangement (later localised to PMP22).
Molecular Genetics & Neuropathology of CMT1A
- Reviewed the chromosome 17 CMT1A locus and the PMP22 gene-dosage mechanism.
- Correlated the CMT1A duplication with sural nerve onion-bulb pathology.
- Linked the Trembler mouse PMP22 point mutations to the human phenotype.
Population Genetic Diagnosis of CMT (Next-Generation Sequencing)
- Population-based sample of 81 CMT families analysed with a 32-gene NGS panel.
- PMP22 duplication was the single most common mutation (11 of 81 families, 14%); a pathogenic mutation was found in 46% overall.
- Mutations were also found in non-classical CMT genes, supporting broad panels over single-gene testing.
Spine Deformity as a Hallmark of CMT4C
- SH3TC2 mutations cause autosomal-recessive demyelinating CMT4C.
- Scoliosis or kyphoscoliosis and foot deformities were present in almost all patients and were often the presenting feature.
- Recommended neurological evaluation when scoliosis is found in this context.
Prevalence of the PMP22 Genomic Disorder
- Methodology study estimating the population prevalence of recurrent CNV genomic disorders mediated by low-copy-repeat nonallelic homologous recombination.
- Includes the PMP22 (17p11.2) duplication/deletion - the molecular basis of CMT1A and HNPP - among the calibration disorders with previously established prevalence.
- Newly estimated prevalences for the 1q21.1, 15q13.3 and 16p11.2 CNV syndromes using a linear-regression model.
Coleman Block Test (Classic Description)
- Described the lateral block test to assess hindfoot flexibility in the cavovarus foot.
- If hindfoot varus corrects when the 1st ray is off-loaded, the varus is forefoot-driven and flexible.
- Determines whether correction should target the forefoot alone or also the hindfoot.