Over 400 Genetic Bone Disorders | Pattern Recognition | Radiographic Diagnosis
- Achondroplasia - FGFR3 mutation, rhizomelic shortening, champagne-glass pelvis, spinal stenosis
- Osteogenesis imperfecta - collagen type I defect, blue sclerae, fractures, bisphosphonate treatment
- Pattern recognition - look at bone density (decreased/increased) and shape (epiphyseal/metaphyseal/diaphyseal)
- Spinal complications - cervical instability, kyphosis, stenosis are major surgical considerations
- New treatments - vosoritide for achondroplasia, bisphosphonates for OI, enzyme replacement for MPS
- “Achondroplasia: FGFR3 mutation, autosomal dominant, normal intelligence, spinal stenosis risk
- “OI Type I: blue sclerae, hearing loss - Type II is lethal
- “Mucopolysaccharidoses show dysostosis multiplex on X-ray
- “Osteopetrosis: dense but brittle bones - osteoclast dysfunction
FGFR3 mutation causing decreased endochondral ossification. Rhizomelic shortening (proximal limbs), champagne-glass pelvis, frontal bossing, trident hand. Major orthopaedic issues: foramen magnum stenosis, spinal stenosis, genu varum.
Collagen type I defect (COL1A1/COL1A2). Blue sclerae, fragile bones, hearing loss. Type I (mild, blue sclerae) to Type II (lethal). Treatment: bisphosphonates, rodding procedures. Beware NAI misdiagnosis.
Lysosomal storage disorders - enzyme deficiency leads to GAG accumulation. Dysostosis multiplex on X-ray. Hurler (MPS I) most severe, Morquio (MPS IV) has unique skeletal features. Enzyme replacement available for some.
Osteopetrosis - osteoclast dysfunction causing dense but brittle bones. Erlenmeyer flask deformity, sandwich vertebrae. Severe form needs BMT. Pyknodysostosis - dense bones + acroosteolysis (Toulouse-Lautrec had this).
Overview and Epidemiology
Skeletal dysplasias are a heterogeneous group of over 400 genetic disorders affecting bone and cartilage development. While individually rare, they collectively represent a significant burden of genetic skeletal disease with an overall incidence of approximately 1 in 5000 live births. [1,2]
The Nosology and Classification of Genetic Skeletal Disorders (revised periodically by the International Skeletal Dysplasia Society) currently recognizes 461 different conditions organized into 42 groups based on clinical, radiographic, and molecular criteria.
- Disorders of cartilage growth (achondroplasia group)
- Disorders of collagen synthesis (osteogenesis imperfecta)
- Lysosomal storage disorders (mucopolysaccharidoses)
- Disorders of mineral homeostasis (rickets, hypophosphatasia)
- Disorders of bone resorption (osteopetrosis)
Radiographic diagnosis of skeletal dysplasias primarily relies on pattern recognition. Key features to assess: (1) Bone density - decreased (osteopenic) or increased (sclerosing); (2) Which part of the bone is affected - epiphyseal, metaphyseal, or diaphyseal; (3) Limb proportions - rhizomelic (proximal), mesomelic (middle), or acromelic (distal); (4) Spine involvement - platyspondyly, vertebral beaking.
Common Viable Skeletal Dysplasias:
- Achondroplasia (1:25,000) - most common
- Osteogenesis imperfecta (1:15,000)
- Spondyloepiphyseal dysplasia (1:100,000)
- Multiple epiphyseal dysplasia (1:100,000)
- Cleidocranial dysplasia (1:1,000,000)
Pathophysiology
Molecular Basis
Skeletal dysplasias result from mutations affecting various pathways:
- Dysplasia
- Achondroplasia, thanatophoric dysplasia
- Effect
- Gain-of-function inhibits chondrocyte proliferation
- Dysplasia
- Osteogenesis imperfecta
- Effect
- Abnormal type I collagen synthesis
- Dysplasia
- SED congenita, Stickler syndrome
- Effect
- Abnormal type II collagen in cartilage
- Dysplasia
- Mucopolysaccharidoses
- Effect
- GAG accumulation in tissues
- Dysplasia
- Osteopetrosis
- Effect
- Osteoclast dysfunction - no resorption
- Dysplasia
- Cleidocranial dysplasia
- Effect
- Defective intramembranous ossification
Bone Formation Affected
- Achondroplasia group - growth plate dysfunction
- Epiphyseal dysplasias - cartilage model affected
- Most short-limbed dwarfism
- Cleidocranial dysplasia - clavicle, skull
- Affects flat bones
- Osteogenesis imperfecta - affects all type I collagen-containing tissues
- Osteopetrosis - affects bone remodeling throughout skeleton
Classification
Radiographic Classification Approach
- Osteogenesis imperfecta - fragile bones, multiple fractures
- Hypophosphatasia - rickets-like changes, deficient alkaline phosphatase
- Idiopathic juvenile osteoporosis
- Osteopetrosis - diffuse sclerosis, Erlenmeyer flask
- Pyknodysostosis - sclerosis + acroosteolysis
- Osteopoikilosis - spotty sclerosis (benign)
- Melorheostosis - flowing candle wax appearance
Density assessment on radiograph is the first step in classifying an unknown skeletal dysplasia. This immediately narrows the differential diagnosis.
Clinical Presentation
Skeletal dysplasias present with characteristic clinical features that guide initial diagnosis:
General Presentation Patterns
- Proportionate vs disproportionate (most skeletal dysplasias)
- Short limb vs short trunk patterns
- Measure arm span, sitting height, upper/lower segment ratio
- Rhizomelic: Proximal segment shortened (achondroplasia)
- Mesomelic: Middle segment shortened (Langer mesomelic)
- Acromelic: Distal segment shortened (acromicric dysplasia)
- Short trunk: Spine predominantly affected (SED, Morquio)
Specific Clinical Signs
- Dysplasia
- Osteogenesis imperfecta Type I
- Significance
- Thin sclerae showing choroid vessels
- Dysplasia
- Achondroplasia
- Significance
- Large head with prominent forehead
- Dysplasia
- Achondroplasia
- Significance
- Gap between 3rd and 4th fingers
- Dysplasia
- Cleidocranial dysplasia
- Significance
- Absent/hypoplastic clavicles
- Dysplasia
- MPS (Hurler, Hunter)
- Significance
- GAG accumulation in soft tissues
- Dysplasia
- MPS I (Hurler/Scheie)
- Significance
- NOT in Hunter syndrome
History Taking
- Pattern of inheritance (AD, AR, X-linked)
- 80% of achondroplasia cases are new mutations
- Parental age (increased paternal age associated with new dominant mutations)
- Motor milestones - often delayed in severe forms
- Cognitive development - normal in most (except MPS types I-III)
- Growth velocity and pattern
- Respiratory issues (thoracic involvement, obstructive sleep apnoea)
- Hearing loss (OI, MPS)
- Visual problems (corneal clouding in MPS)
- Joint pain/stiffness
Differential Diagnosis
- Pattern / Body Segment
- Rhizomelic short-limb
- Discriminating Feature
- Frontal bossing, trident hand, champagne-glass pelvis
- Confirmatory Test
- FGFR3 G380R mutation
- Pattern / Body Segment
- Mild rhizomelic short-limb
- Discriminating Feature
- Milder than achondroplasia, often normal facies
- Confirmatory Test
- FGFR3 (non-G380R) mutation
- Pattern / Body Segment
- Severe rhizomelic, lethal
- Discriminating Feature
- Narrow thorax, telephone-receiver femora
- Confirmatory Test
- FGFR3 mutation (lethal alleles)
- Pattern / Body Segment
- Short-trunk
- Discriminating Feature
- Platyspondyly plus epiphyseal change, odontoid hypoplasia
- Confirmatory Test
- COL2A1 mutation
- Pattern / Body Segment
- Short-trunk
- Discriminating Feature
- Normal intelligence, dysostosis multiplex, corneal clouding
- Confirmatory Test
- Urine GAG, GALNS enzyme assay
- Pattern / Body Segment
- Short-limb with deformity
- Discriminating Feature
- Blue sclerae, fractures, Wormian bones
- Confirmatory Test
- COL1A1/COL1A2 mutation
- Pattern / Body Segment
- Rhizomelic short-limb
- Discriminating Feature
- Normal facies/skull, marked epiphyseal/metaphyseal change
- Confirmatory Test
- COMP mutation
- Pattern / Body Segment
- Disproportionate, acquired
- Discriminating Feature
- Metaphyseal fraying/cupping, bowing, low/normal Ca
- Confirmatory Test
- ALP, phosphate, vitamin D, PHEX
Investigations
Radiographic Assessment
The skeletal survey is the cornerstone of diagnosis in skeletal dysplasias:
- Skull (AP, lateral)
- Spine (AP, lateral)
- Chest (AP)
- Pelvis (AP)
- Long bones (humerus, radius/ulna, femur, tibia/fibula)
- Hand (AP)
-
Bone Density Assessment
- Decreased: OI, hypophosphatasia
- Increased: osteopetrosis, pyknodysostosis
-
Location of Involvement
- Epiphyseal: MED, SED, chondrodysplasia punctata
- Metaphyseal: achondroplasia, rickets, metaphyseal chondrodysplasia
- Diaphyseal: progressive diaphyseal dysplasia, melorheostosis
-
Limb Proportions
- Measure humerus and femur lengths
- Calculate rhizomelic ratio
-
Spine Evaluation
- Vertebral height and shape
- Interpedicular distance
- Platyspondyly, beaking, stenosis
Genetic Testing
- Confirm clinical/radiographic diagnosis
- Genetic counselling
- Prenatal diagnosis in subsequent pregnancies
- Access to specific treatments
- Single gene testing (when diagnosis clear)
- Skeletal dysplasia gene panels (50-500 genes)
- Exome/genome sequencing (atypical presentations)
Laboratory Tests
- Bone markers, vitamin D, calcium
- Collagen analysis (skin biopsy, historical)
- Genetic testing (COL1A1/COL1A2)
- Urine GAG screening (dermatan sulfate, heparan sulfate)
- Enzyme assay in leukocytes (specific enzyme for each MPS type)
- Genetic confirmation
- Alkaline phosphatase (low in hypophosphatasia)
- Calcium, phosphate, vitamin D
- PTH levels
Additional Imaging
- Foramen magnum stenosis assessment (achondroplasia)
- Spinal stenosis evaluation
- Cervical cord compression
- Brain MRI if developmental concerns
- 3D reconstruction for surgical planning
- Cervical spine assessment
- Cardiac involvement in MPS
- Aortic root in Marfan syndrome (connective tissue, not dysplasia)
Antenatal Diagnosis and Prediction of Lethality
Many skeletal dysplasias are first detected on routine obstetric ultrasound, and the structured antenatal assessment - especially the prediction of lethality - is examinable.
Sonographic workup: measure all long bones (a femur length below roughly the 5th centile or markedly short for gestation flags a dysplasia), define the limb-segment pattern (rhizo-/meso-/acromelic), assess bone mineralisation (a poorly ossified skull/spine suggests OI or hypophosphatasia), look for fractures, bowing and angulation, examine the hands and feet (polydactyly), and assess the thorax.
Predicting lethality is the critical decision, because death is driven by pulmonary hypoplasia from a small chest, not by the limb shortening. Features predicting a lethal outcome include a small thoracic circumference, a thoracic-to-abdominal circumference ratio below about 0.6, a femur-length-to-abdominal-circumference ratio below about 0.16, and a narrow "bell-shaped" chest with short ribs.
thanatophoric dysplasia (the commonest lethal form, FGFR3, "telephone-receiver" femora and a narrow thorax), osteogenesis imperfecta type II (multiple intrauterine fractures, beaded ribs, a poorly mineralised calvarium), achondrogenesis (severe under-mineralisation), and campomelic dysplasia (bowed long bones, with XY sex reversal).
targeted fetal molecular testing (amniocentesis or CVS with FGFR3/COL1 panels), 3D ultrasound or low-dose fetal CT of the skeleton, and multidisciplinary genetic counselling - the diagnosis guides delivery planning and perinatal palliative care for lethal forms. Exam point: in a fetus with short limbs, measure the chest - thoracic size predicts survival.

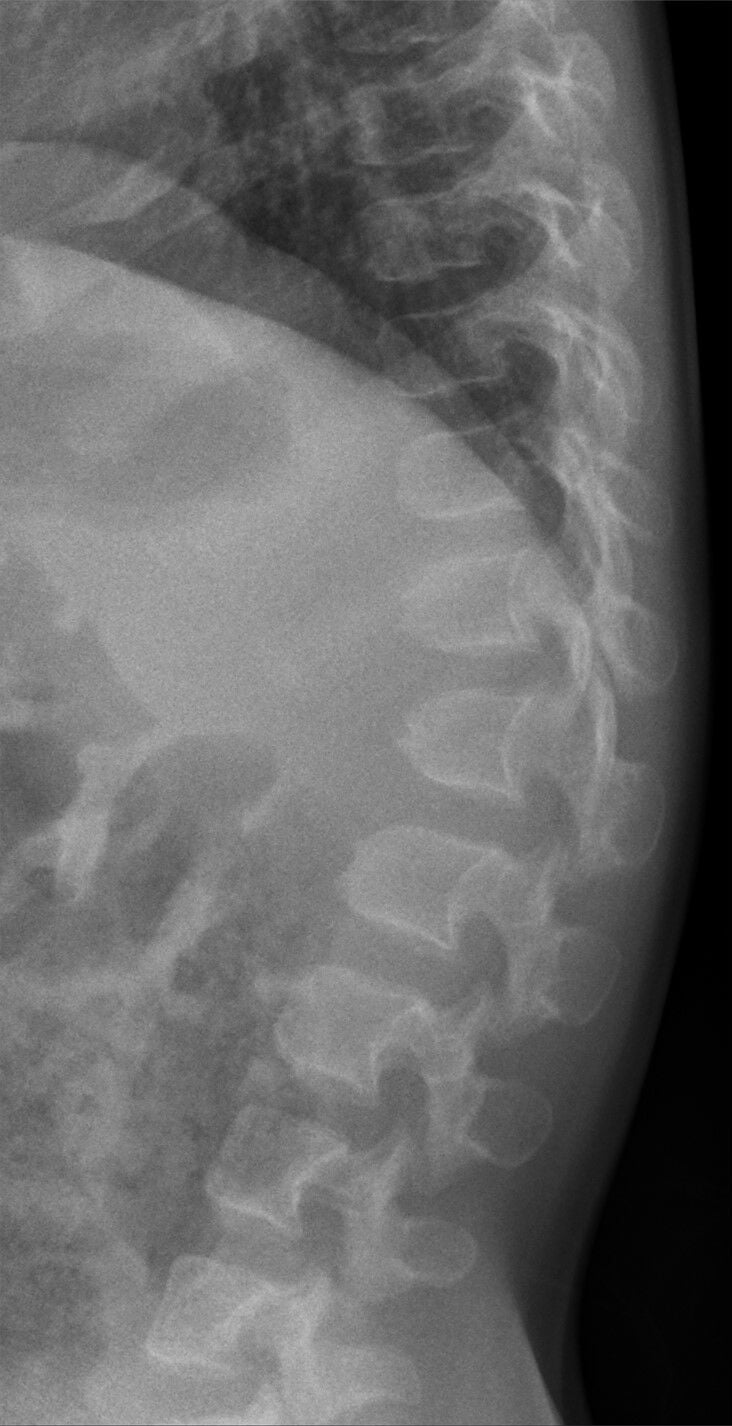

Achondroplasia
Achondroplasia is the most common viable skeletal dysplasia, caused by gain-of-function mutations in the FGFR3 gene. [3,4]
Genetics and Pathophysiology
- Gene: FGFR3 (fibroblast growth factor receptor 3) on chromosome 4p16.3
- Mutation: G380R (glycine to arginine) in 97% of cases
- Inheritance: Autosomal dominant, but 80% are new mutations
- Mechanism: Constitutive activation of FGFR3 inhibits chondrocyte proliferation in growth plates
FGFR3 mutations cause a spectrum of severity: Thanatophoric dysplasia (most severe, lethal) through SADDAN syndrome to Achondroplasia to Hypochondroplasia (mildest). All are gain-of-function mutations with varying degrees of receptor activation.
Clinical Features
- Macrocephaly with frontal bossing
- Midface hypoplasia
- Depressed nasal bridge
- Normal intelligence
- Rhizomelic shortening (proximal limbs most affected)
- Trident hand configuration
- Genu varum (tibial bowing)
- Thoracolumbar kyphosis (in infancy)
- Exaggerated lumbar lordosis (develops later)
Radiographic Features
- Finding
- Enlarged calvarium, small skull base, foramen magnum stenosis
- Clinical Significance
- Risk of cervicomedullary compression
- Finding
- Narrow interpedicular distance caudally, short pedicles, small canal
- Clinical Significance
- Spinal stenosis - major cause of morbidity
- Finding
- Champagne-glass shape, horizontal acetabular roofs, narrow sciatic notch
- Clinical Significance
- Characteristic diagnostic feature
- Finding
- Rhizomelic shortening, metaphyseal flaring, chevron deformity of femur
- Clinical Significance
- Affects endochondral ossification
- Finding
- Trident configuration, short tubular bones
- Clinical Significance
- Persistent gap between 3rd and 4th fingers
Orthopaedic Management
- Foramen magnum stenosis - monitor in infancy, decompression if symptomatic
- Thoracolumbar kyphosis - bracing, fusion if progressive
- Spinal stenosis - develops in adults, may need multilevel decompression
- Genu varum - guided growth or osteotomy
- Limb lengthening - controversial, significant complications
- Avoid obesity to reduce mechanical stress
- Vosoritide (C-natriuretic peptide analogue) - FDA approved 2021
- Works by antagonizing FGFR3 signaling
- Shown to increase growth velocity in clinical trials
Osteogenesis Imperfecta
Osteogenesis imperfecta (OI) is a group of genetic disorders affecting type I collagen, resulting in bone fragility and other connective tissue manifestations. [5,6]
Genetics
- Genes: Primarily COL1A1 and COL1A2 (encode type I collagen)
- Inheritance: Mostly autosomal dominant (Types I-IV)
- Newer genes: CRTAP, LEPRE1, PPIB (recessive forms)
Sillence Classification
- Severity
- Mild
- Sclerae
- Blue
- Key Features
- Fractures after walking, normal stature, hearing loss common
- Severity
- Lethal
- Sclerae
- Dark blue
- Key Features
- Multiple intrauterine fractures, beaded ribs, perinatal death
- Severity
- Severe
- Sclerae
- Variable
- Key Features
- Progressive deformity, short stature, wheelchair by teens
- Severity
- Moderate
- Sclerae
- White/blue
- Key Features
- Moderate fragility, normal sclerae in adults, dentinogenesis imperfecta
Clinical Features
- Blue sclerae (thin sclerae showing choroidal vessels)
- Bone fragility (multiple fractures)
- Hearing loss (conductive then sensorineural)
- Dentinogenesis imperfecta (opalescent teeth)
- Joint hypermobility
- Easy bruising
- Short stature (severe forms)
- Basilar invagination (Type III)
OI can be mistaken for non-accidental injury (NAI) due to multiple unexplained fractures. Key differentiators: blue sclerae, family history, Wormian bones on skull X-ray, osteopenia, and genetic testing. Always consider OI before diagnosing NAI in an infant with fractures.
Radiographic Features
- Generalized osteopenia
- Multiple fractures at various stages of healing
- Wormian bones (multiple small bones in skull sutures)
- Codfish vertebrae (biconcave)
- Gracile long bones
- Popcorn calcification in epiphyses (severe forms)
Management
- Bisphosphonates (pamidronate, zoledronic acid) - increase BMD, reduce fractures
- Calcium and vitamin D supplementation
- Physical therapy - strengthen muscles, prevent falls
- Intramedullary rodding - stabilize long bones, prevent deformity
- Telescoping rods (Bailey-Dubow, Fassier-Duval) - grow with child
- Spinal fusion for scoliosis
- Basilar impression decompression if needed
Intramedullary rodding in OI uses telescoping designs that elongate with growth. Key principles: rod both femurs and tibias for walking patients, use solid rods for non-ambulatory patients, fixation should span entire bone, and augment with bisphosphonates pre- and post-operatively.
Mucopolysaccharidoses
The mucopolysaccharidoses (MPS) are lysosomal storage disorders caused by deficiency of enzymes that degrade glycosaminoglycans (GAGs). The accumulation of GAGs in tissues causes progressive multisystem disease. [7]
Classification
- Name
- Hurler/Scheie
- Enzyme Deficiency
- Alpha-L-iduronidase
- Key Features
- Most severe, cognitive decline, corneal clouding
- Name
- Hunter
- Enzyme Deficiency
- Iduronate sulfatase
- Key Features
- X-linked, no corneal clouding, variable severity
- Name
- Sanfilippo
- Enzyme Deficiency
- Various heparan sulfate enzymes
- Key Features
- Behavioral issues, mild skeletal involvement
- Name
- Morquio
- Enzyme Deficiency
- Galactose-6-sulfatase (A) or B-galactosidase (B)
- Key Features
- Normal intelligence, severe skeletal involvement, odontoid hypoplasia
- Name
- Maroteaux-Lamy
- Enzyme Deficiency
- Arylsulfatase B
- Key Features
- Normal intelligence, severe skeletal involvement
Dysostosis Multiplex
The radiographic constellation of findings in MPS is termed "dysostosis multiplex":
- J-shaped sella turcica
- Paddle-shaped (oar-shaped) ribs
- Hook-shaped vertebrae with anteroinferior beaking
- Diaphyseal widening of long bones
- Bullet-shaped metacarpals with proximal pointing
- Hypoplastic L1/L2 vertebra causing kyphosis
Orthopaedic Considerations
- Odontoid hypoplasia (especially Morquio)
- Atlantoaxial instability
- Cervical stenosis
- May need occipitocervical fusion
- Gibbus deformity at thoracolumbar junction
- Progressive kyphosis
- May need spinal fusion
- Genu valgum (especially Morquio)
- Hip dysplasia
- Guided growth or osteotomy
Treatment
- Enzyme replacement therapy (ERT) - available for MPS I, II, IVA, VI
- Hematopoietic stem cell transplant - best if early, for MPS I
- Gene therapy - under investigation
- Address spinal instability early
- Joint procedures as needed
- Cardiac valve surgery in some types
Morquio syndrome (MPS IVA) has unique features: normal intelligence, severe skeletal involvement, odontoid hypoplasia with atlantoaxial instability. These patients need cervical spine precautions for any anaesthesia. Always obtain flexion-extension cervical spine imaging before surgery.
Other Important Dysplasias
- Osteoclast dysfunction - failure of bone resorption
- Diffuse sclerosis but paradoxically brittle bones
- Erlenmeyer flask deformity (failure of metaphyseal remodeling)
- Sandwich vertebrae, bone-within-bone appearance
- Severe form: pancytopenia, cranial nerve compression
- Treatment: BMT for severe infantile form
- Cathepsin K deficiency
- Dense bones + acroosteolysis (terminal phalangeal resorption)
- Open fontanelles, micrognathia
- Toulouse-Lautrec reportedly had this condition
- Fractures common despite dense appearance
Sclerosing dysplasias paradoxically have fragile bones despite increased radiographic density due to abnormal bone remodeling.
A classic pattern-recognition dysplasia this topic must describe rather than just name - examiners reward the pentad of signs. Diastrophic dysplasia is autosomal recessive, caused by mutations in SLC26A2 (DTDST), the sulfate transporter, producing undersulfated cartilage proteoglycan; intelligence is normal and the dwarfism is short-limbed (rhizomelic).
- "Hitchhiker thumb" - a proximally-set, abducted, hypermobile thumb with a short first metacarpal.
- "Cauliflower ear" - acute cystic swelling of the pinna in infancy that later calcifies/ossifies.
- Rigid, severe equinovarus (clubfoot) that resists casting.
- Cervical kyphosis - the key spinal danger, which can be progressive with cord-compression risk and may need early posterior fusion.
- Multiple joint contractures and dislocations (symphalangism, "gull-wing" interphalangeal deformity), with progressive scoliosis, early osteoarthritis and a cleft palate in many.
monitor and treat the cervical kyphosis, manage the recurrent/resistant clubfoot and contractures, address progressive scoliosis, and anticipate early joint degeneration. (Contrast with pseudoachondroplasia - COMP mutation, normal face/skull, normal at birth then growth failure around age two, marked epiphyseal/metaphyseal change, ligamentous laxity and C1-C2 instability.)
Management
Management of skeletal dysplasias requires a multidisciplinary approach addressing both medical and orthopaedic needs.
- Vosoritide (achondroplasia) - C-natriuretic peptide analogue, FDA approved 2021
- Bisphosphonates (OI) - pamidronate, zoledronic acid to increase BMD
- Enzyme replacement (MPS) - laronidase (MPS I), idursulfase (MPS II), elosulfase (MPS IVA)
- HSCT (MPS I severe) - best outcomes if performed early
- Growth hormone - not effective in most true skeletal dysplasias
- Vitamin D and calcium supplementation
- Respiratory support - CPAP for obstructive sleep apnoea
- Pain management - multimodal approach
- Regular growth measurements
- Sleep studies for at-risk patients
- Cardiac echo for MPS patients
- Hearing and vision screening
The medical management aims to optimize quality of life and address systemic manifestations.
Surgical Technique
Intramedullary Rodding in OI
- Recurrent fractures (greater than 2 per year in same bone)
- Progressive bowing deformity
- Anticipated fracture through osteopenic segment
- Femur and tibia most commonly rodded
- Correct angular deformity with osteotomies if needed
- Rod should span entire bone length
- Avoid stress risers at rod ends
- Combine with bisphosphonate therapy
- Mechanism
- Fixed length
- Best For
- Non-ambulatory patients
- Consideration
- Needs replacement with growth
- Mechanism
- Telescopes at metaphysis
- Best For
- Growing children
- Consideration
- Complex, can malfunction
- Mechanism
- Telescopes at diaphysis
- Best For
- Growing children
- Consideration
- Newer design, better function
Deformity Correction Osteotomies
Technique:
- Multiple osteotomies through apex of deformity
- "Shish-kebab" technique - threading fragments on rod
- Minimal periosteal stripping to preserve blood supply
Complications
Orthopaedic Complications
- Foramen magnum stenosis (achondroplasia) - brainstem compression
- Spinal stenosis - progressive neurological deficit
- Atlantoaxial instability (MPS, SED) - risk of cord injury
- Cervical kyphosis (diastrophic dysplasia) - may need early fusion
- Basilar invagination (OI) - cranial settling
- Progressive scoliosis - respiratory compromise
- Angular deformity progression - genu varum/valgum
- Premature osteoarthritis (MED, SED)
- Pathological fractures (OI, osteopetrosis paradoxically)
- Hip dysplasia and dislocation (MPS, congenital dysplasias)
- Growth plate injury from recurrent fractures
- Early-onset degenerative disease
- Joint contractures (MPS)
- Ligamentous laxity (OI, Ehlers-Danlos overlap)
Medical Complications
- Complication
- Obstructive sleep apnoea, obesity
- Management
- Sleep study, CPAP, weight management
- Complication
- Hearing loss, respiratory failure (severe)
- Management
- Audiology, pulmonology input
- Complication
- Cardiac valve disease, cognitive decline
- Management
- Echo surveillance, HSCT if eligible
- Complication
- Pancytopenia, blindness, deafness
- Management
- BMT, supportive care
- Complication
- Respiratory failure (lethal)
- Management
- Perinatal palliative care
Surgical Complications
- OI: Implant cutout, refracture at implant ends
- Osteopetrosis: Difficult drilling, delayed healing
- Solution: Telescoping rods in OI, staged treatment
- Dural ectasia (some syndromes)
- Abnormal anatomy
- Difficult intubation (short neck, atlantoaxial instability)
- Increased bleeding risk
Cervical spine instability: Morquio, SED, Down syndrome - fibreoptic intubation may be needed. Difficult airway: short neck, large tongue, restricted mouth opening (MPS). Always have experienced anaesthesia team for skeletal dysplasia patients.
Postoperative Care
General Postoperative Principles
- Close neurological monitoring after spinal procedures
- Pain management - multimodal approach
- Early mobilization when safe
- DVT prophylaxis (mechanical, consider chemical in adults)
- Protected weight bearing initially
- Serial radiographs to confirm healing
- Monitor for implant complications
- Resume bisphosphonates when appropriate
Rehabilitation
- Mobilization
- Day 1-2
- Weight Bearing
- PWB 6 weeks, WBAT 12 weeks
- Return to Activity
- 4-6 months
- Mobilization
- Day 1-2
- Weight Bearing
- PWB 6 weeks, WBAT 12 weeks
- Return to Activity
- 4-6 months
- Mobilization
- Collar 6-12 weeks
- Weight Bearing
- N/A
- Return to Activity
- 3-6 months
- Mobilization
- Day 1
- Weight Bearing
- Full
- Return to Activity
- 6-12 weeks
Outcomes
Outcomes by Condition
- Life expectancy near normal with appropriate management
- Foramen magnum decompression highly effective when needed
- Spinal stenosis surgery improves quality of life
- Vosoritide showing promise for improved height outcomes
- Bisphosphonates: 30-50% reduction in fracture rate
- Rodding: 90%+ reduction in fractures in rodded bones
- Quality of life significantly improved with modern management
- Type II remains uniformly lethal
- Success Rate
- 85-95%
- Complications
- Rod migration, refracture
- Revision Rate
- 20-30% (growth)
- Success Rate
- 90%+
- Complications
- CSF leak, infection
- Revision Rate
- 5-10%
- Success Rate
- 80-90%
- Complications
- Nonunion, progression
- Revision Rate
- 10-15%
- Success Rate
- 70-80%
- Complications
- Instability, recurrence
- Revision Rate
- 15-20%
Guidelines, Registries & Global Practice
Global Epidemiology
Skeletal dysplasias are individually rare but collectively important. Population-based data from the Utah Birth Defect Network give a birth prevalence of about 3.0 per 10,000 births (rising to 20 per 10,000 stillbirths), with osteogenesis imperfecta (0.79/10,000), thanatophoric dysplasia (0.43/10,000) and achondroplasia (0.35/10,000) the commonest categories. According to PubMed, see Stevenson DA et al. Am J Med Genet A 2012 (DOI). The International Skeletal Dysplasia Society 2019 Nosology recognises 461 disorders in 42 groups, with a causal gene now identified for 92% — Mortier GR et al. Am J Med Genet A 2019 (DOI). Achondroplasia (the commonest non-lethal dysplasia, roughly 1:25,000-1:30,000) and OI (roughly 1:15,000-1:20,000) account for the bulk of the orthopaedic caseload worldwide; incidence is broadly uniform across populations because most cases of achondroplasia arise from recurrent de novo FGFR3 G380R mutations linked to advanced paternal age.
Guidance Across Jurisdictions
- Scope
- Nosology & diagnosis
- Key Position
- 461-disorder nosology; radiographic group then targeted molecular testing
- Scope
- Achondroplasia lifetime care
- Key Position
- Structured foramen-magnum surveillance in infancy; multidisciplinary follow-up
- Scope
- Vosoritide & ERT commissioning
- Key Position
- Highly specialised commissioning with defined eligibility and stopping rules
- Scope
- Drug approval
- Key Position
- Vosoritide approved (FDA 2021, EMA 2021, TGA 2022); ERT approved for MPS I/II/IVA/VI
- Scope
- Orthopaedic surgery in MPS
- Key Position
- Early cervical assessment; flexion-extension imaging before any anaesthesia
Registry & Disease-Specific Networks
- CLARITY / international OI registries and the Brittle Bone Disease Consortium collect longitudinal fracture, bisphosphonate and rodding outcome data.
- Achondroplasia natural-history and post-marketing registries (e.g. the lifeline / industry-sponsored vosoritide cohorts) track growth, foramen magnum events and spinal stenosis.
- MPS patient registries (Hunter Outcome Survey, MPS I Registry) inform ERT eligibility, cardiac and skeletal surveillance, and long-term safety.
Practice Variation & Access
- Limb lengthening is performed selectively and is more common in parts of Europe and the Middle East than in the UK/Australasia, where it remains controversial.
- Vosoritide funding varies widely: reimbursed in several European systems and through specialised commissioning in the UK, but access elsewhere is often via managed-access or patient programmes.
- HSCT vs ERT sequencing for severe MPS I differs by centre and timing of diagnosis (newborn screening availability).
Across FRACS, FRCS (Tr & Orth) and international fellowship exams you must recognise common skeletal dysplasias radiographically, understand the orthopaedic complications of achondroplasia and OI, know the principles of telescoping-rod fixation in OI, and recognise cervical instability risks in MPS/SED. Classification by bone density and location is a common viva framework.
MCQ Practice Points
Q: What is the most common skeletal dysplasia and its genetic basis? A: Achondroplasia, caused by gain-of-function mutation in FGFR3 (fibroblast growth factor receptor 3). This mutation inhibits chondrocyte proliferation in the growth plate, causing rhizomelic (proximal) limb shortening with normal trunk length.
Q: What is the life-threatening complication of achondroplasia in infancy? A: Foramen magnum stenosis causing cervicomedullary compression. The small foramen magnum combined with atlantoaxial instability can cause brainstem compression, central apnea, and sudden death. MRI screening is recommended in the first 2 years.
Q: How do you distinguish spondyloepiphyseal dysplasia (SED) from multiple epiphyseal dysplasia (MED)? A: SED has short trunk with vertebral involvement (platyspondyly) plus epiphyseal abnormalities. MED has normal trunk height with only epiphyseal involvement. Both cause premature osteoarthritis, but SED patients are shorter and have spinal deformity.
Q: What orthopaedic complications are common in achondroplasia? A: Spinal stenosis (lumbar, may need multilevel decompression), foramen magnum stenosis (cervical), thoracolumbar kyphosis (often self-corrects), genu varum (tibial bowing), and atlantoaxial instability. Hip and knee arthroplasty may be challenging due to anatomic variants.
At a Glance
Skeletal dysplasias encompass 400+ genetic bone disorders (collective incidence ~1:5,000) classified by radiographic pattern: osteopenic (OI), sclerosing (osteopetrosis), short-limbed (achondroplasia), or short-trunk (SED, Morquio). Achondroplasia (most common viable dysplasia, 1:25,000) results from FGFR3 gain-of-function mutation causing rhizomelic shortening, champagne-glass pelvis, trident hand, and progressive spinal stenosis. Osteogenesis imperfecta (COL1A1/COL1A2 collagen type I defects) presents with blue sclerae, fractures, hearing loss, and dentinogenesis imperfecta—treated with bisphosphonates and rodding procedures. Mucopolysaccharidoses (lysosomal storage disorders) show dysostosis multiplex on X-ray (J-shaped sella, hook vertebrae, paddle ribs). Spinal complications (cervical instability, stenosis, kyphosis) are major surgical considerations across dysplasia types. Newer treatments include vosoritide for achondroplasia and enzyme replacement for MPS.
STAMPAchondroplasia Features - STAMP
Hook:STAMP the diagnosis - achondroplasia leaves its STAMP on the skeleton
BONEDOsteogenesis Imperfecta Features - BONED
Hook:BONED - brittle bones need this easy mnemonic
JDHIPDysostosis Multiplex Features - JDHIP
Hook:JDHIP - J-shaped sella, Dysostosis, Hook vertebrae = MPS storage diseases
Clinical Decision Scenarios
Practise clinical reasoning and management decisions out loud
“Classic presentation of achondroplasia or related FGFR3 disorder. The examiner wants systematic evaluation.”
“Important scenario - need to distinguish OI from NAI while ensuring child safety.”
“Tests knowledge of MPS complications and perioperative considerations.”
“Tests systematic approach to pattern recognition in skeletal dysplasias.”
“Tests knowledge of evidence-based management in skeletal dysplasia.”
CLASSIFICATION APPROACH
- Bone DENSITY first: osteopenic vs sclerosing
- LOCATION: epiphyseal, metaphyseal, or diaphyseal
- LIMB proportions: rhizomelic, mesomelic, acromelic
- SPINE: platyspondyly, beaking, canal size
ACHONDROPLASIA - STAMP
- S = Spinal stenosis (narrow canal, short pedicles)
- T = Trident hand (gap between 3rd and 4th fingers)
- A = Autosomal dominant, FGFR3 gain-of-function
- M = Macrocephaly, frontal bossing, midface hypoplasia
- P = Pelvis champagne-glass shaped
OSTEOGENESIS IMPERFECTA - BONED
- B = Blue sclerae (Type I)
- O = Osteopenia, multiple fractures
- N = Normal intelligence (distinguish from NAI)
- E = Ear - hearing loss (otosclerosis)
- D = Dentinogenesis imperfecta
OI TYPES
- Type I = Mild, blue sclerae, hearing loss - MOST COMMON
- Type II = Lethal, multiple intrauterine fractures
- Type III = Severe, progressive deformity, wheelchair
- Type IV = Moderate, white/normal sclerae in adults
DYSOSTOSIS MULTIPLEX (MPS)
- J-shaped sella turcica
- Paddle-shaped (oar) ribs
- Hook vertebrae with anteroinferior beaking
- Diaphyseal widening of long bones
- Bullet metacarpals with proximal pointing
MORQUIO SYNDROME (MPS IVA)
- NORMAL INTELLIGENCE - key differentiator
- Severe skeletal involvement
- Odontoid hypoplasia - cervical instability
- MUST get flex-ext C-spine before any anaesthesia
- ERT available (elosulfase alfa)
TREATMENT
- Achondroplasia: vosoritide (C-natriuretic peptide analogue) - FDA 2021
- OI: bisphosphonates (increase BMD), rodding procedures
- MPS: ERT for Types I, II, IVA, VI; BMT for Type I
- Spinal surgery for instability/stenosis
- Limb surgery for deformity correction
Evidence Base
- First disease-modifying therapy for achondroplasia (C-natriuretic peptide analogue)
- 1.57 cm/year increase in annualised growth velocity over placebo
- Final adult-height impact and long-term harms remain unknown
- Bisphosphonates increase BMD in children and adults with OI
- Fracture-reduction evidence is inconsistent and not conclusive
- Optimal agent, dose, duration and long-term safety remain undefined
- ERT improves respiratory function and physical capacity in MPS I
- No reversal of established skeletal (dysostosis multiplex) changes
- Supports early treatment before irreversible bony deformity
- Collective birth prevalence of skeletal dysplasias ~3 per 10,000
- OI and the FGFR3 chondrodysplasia group are the largest diagnostic categories
- Lethal forms cluster in stillbirths and terminations, biasing live-birth figures
- Original delineation of OI Types I-IV (Sillence classification)
- Type I: autosomal dominant, blue sclerae, presenile hearing loss
- Perinatal-lethal group (Type II) with crumpled femora and beaded ribs
- 461 recognised skeletal dysplasias in 42 clinical/molecular groups
- A causal gene is now known for 92% of entities
- Reference framework for diagnosis and research in skeletal dysplasia
Suggested Reading
- Mortier GR, Cohn DH, Cormier-Daire V, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019;179(12):2393-2419. doi:10.1002/ajmg.a.61366
- Krakow D, Rimoin DL. The skeletal dysplasias. Genet Med. 2010;12(6):327-341. doi:10.1097/GIM.0b013e3181daae9b
- Horton WA, Hall JG, Hecht JT. Achondroplasia. Lancet. 2007;370(9582):162-172. doi:10.1016/S0140-6736(07)61090-3
- Savarirayan R, Tofts L, Irving M, et al. Once-daily, subcutaneous vosoritide therapy in children with achondroplasia: a randomised, double-blind, phase 3, placebo-controlled, multicentre trial. Lancet. 2020;396(10252):684-692. doi:10.1016/S0140-6736(20)31541-5
- Marini JC, Forlino A, Bachinger HP, et al. Osteogenesis imperfecta. Nat Rev Dis Primers. 2017;3:17052. doi:10.1038/nrdp.2017.52
- Dwan K, Phillipi CA, Steiner RD, Basel D, Cochrane Cystic Fibrosis and Genetic Disorders Group. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev. 2016;10:CD005088. doi:10.1002/14651858.CD005088.pub4
- Muenzer J. Overview of the mucopolysaccharidoses. Rheumatology (Oxford). 2011;50 Suppl 5:v4-12. doi:10.1093/rheumatology/ker394
- Rajeshwar N, Behr S, Engel N. A primer on skeletal dysplasias. Emerg Radiol. 2022;29(2):385-408. doi:10.1007/s10140-021-02006-4
- Cheung MS, Glorieux FH. Osteogenesis imperfecta: update on presentation and management. Rev Endocr Metab Disord. 2008;9(2):153-160. doi:10.1007/s11154-008-9074-4
- White KK, Bober MB, Engel N, et al. Practical approach to the orthopedic surgical management of the mucopolysaccharidoses. Mol Genet Metab. 2017;122S:142-149. doi:10.1016/j.ymgme.2017.09.006
- Ireland PJ, Pacey V, Zankl A, et al. Optimal management of complications associated with achondroplasia. Appl Clin Genet. 2014;7:117-125. doi:10.2147/TACG.S51485
- Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16(2):101-116. doi:10.1136/jmg.16.2.101
- Wraith JE, Clarke LA, Beck M, et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J Pediatr. 2004;144(5):581-588. doi:10.1016/j.jpeds.2004.01.046
- Panda A, Gamanagatti S, Jana M, Gupta AK. Skeletal dysplasias: A radiographic approach and review of common non-lethal skeletal dysplasias. World J Radiol. 2014;6(10):808-825. doi:10.4329/wjr.v6.i10.808
- Shapiro JR, Germain-Lee EL. Osteogenesis imperfecta: effecting the transition from adolescent to adult medical care. J Musculoskelet Neuronal Interact. 2012;12(1):24-27.
Key Guidelines
- International Skeletal Dysplasia Society Nosology 2019
- AAOS/SRS Guidelines on Spinal Management in Skeletal Dysplasias
Additional Reading
- Spranger JW, Brill PW, Superti-Furga A, et al. Bone Dysplasias: An Atlas of Genetic Disorders of Skeletal Development. 4th ed. Oxford University Press; 2018.
- Bonafe L, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015;167A(12):2869-2892.