Mechanisms, Resistance and Biofilm in Orthopaedics
- Antibiotics are grouped by their TARGET: CELL-WALL synthesis (BETA-LACTAMS - penicillins/cephalosporins/carbapenems inhibit penicillin-binding proteins; GLYCOPEPTIDES like vancomycin bind D-ala-D-ala); PROTEIN synthesis at the ribosome (AMINOGLYCOSIDES, MACROLIDES, CLINDAMYCIN, LINEZOLID, TETRACYCLINES); DNA/RNA (FLUOROQUINOLONES inhibit DNA gyrase, RIFAMPICIN inhibits RNA polymerase, metronidazole); and FOLATE synthesis (trimethoprim-sulfamethoxazole).
- Antibiotics are BACTERICIDAL (kill, e.g. beta-lactams, glycopeptides, aminoglycosides, fluoroquinolones, rifampicin) or BACTERIOSTATIC (inhibit growth, e.g. tetracyclines, macrolides, clindamycin, linezolid); killing is either CONCENTRATION-DEPENDENT (aminoglycosides, fluoroquinolones - high peak matters) or TIME-DEPENDENT (beta-lactams, glycopeptides - time above MIC matters), which guides dosing.
- RESISTANCE arises by four mechanisms: (1) ENZYMATIC INACTIVATION (beta-lactamases, including ESBL and carbapenemases), (2) TARGET MODIFICATION (altered penicillin-binding protein PBP2a in MRSA via mecA; altered D-ala-D-lac in VRE via vanA; gyrase/ribosomal mutations), (3) REDUCED UPTAKE/decreased permeability (porin loss) and (4) EFFLUX PUMPS; resistance genes spread by plasmids/transposons (horizontal gene transfer), producing MRSA, VRE, ESBL and multidrug-resistant gram-negatives.
- BIOFILM is the key orthopaedic concept: bacteria on an implant or dead bone form a structured community embedded in a self-produced extracellular matrix (glycocalyx), which confers antibiotic TOLERANCE (distinct from genetic resistance) through REDUCED antibiotic PENETRATION, SLOW-GROWING/dormant PERSISTER cells and SMALL-COLONY VARIANTS, and an altered microenvironment.
- Because of biofilm tolerance, ANTIBIOTICS ALONE typically FAIL against implant-associated infection (periprosthetic joint infection, implant-related osteomyelitis) - effective treatment requires SURGICAL removal/debridement of the biofilm (and often the implant) PLUS antibiotics; this is why source control is paramount.
- RIFAMPICIN is special: it penetrates the STAPHYLOCOCCAL biofilm and intracellular bacteria well, so it is a key agent for staphylococcal implant infection - but it must ALWAYS be used in COMBINATION (never as monotherapy) because resistance to rifampicin develops rapidly; fluoroquinolones (good bone penetration) are common rifampicin partners for susceptible gram-negative/staph PJI.
- “Targets: cell wall (beta-lactams, vancomycin), ribosome (aminoglycosides/macrolides/clindamycin/linezolid/tetracyclines), DNA/RNA (fluoroquinolones, rifampicin), folate (TMP-SMX).
- “Resistance: enzyme (beta-lactamase), target modification (MRSA PBP2a/mecA, VRE vanA), efflux, reduced uptake; spread by plasmids.
- “Biofilm = TOLERANCE (not resistance): reduced penetration + persisters/small-colony variants -> antibiotics alone fail -> need SURGERY + antibiotics. Rifampicin penetrates staph biofilm (ALWAYS combination).
Bacteria in an extracellular matrix on the implant: reduced antibiotic penetration + persister cells / small-colony variants -> antibiotics alone fail (tolerance, not classic resistance).
Surgical debridement / implant removal (source control) plus antibiotics; rifampicin for staphylococcal biofilm (always in combination).
Antibiotic Classes & Mechanisms
Antibiotics work on four main bacterial targets. Cell-wall synthesis is inhibited by beta-lactams (penicillins, cephalosporins, carbapenems - which block penicillin-binding proteins/transpeptidase) and glycopeptides (vancomycin, teicoplanin - which bind the D-ala-D-ala terminus; key for gram-positives and MRSA). Protein synthesis at the ribosome is inhibited by aminoglycosides (gentamicin - 30S, bactericidal, used in cement/beads), macrolides, clindamycin, linezolid (for MRSA/VRE) and tetracyclines. DNA/RNA is targeted by fluoroquinolones (DNA gyrase - good bone penetration, oral), rifampicin (RNA polymerase - excellent biofilm/intracellular penetration) and metronidazole (anaerobes). Folate synthesis is inhibited by trimethoprim-sulfamethoxazole. Two further agents matter in resistant gram-positive infection: daptomycin, a cyclic lipopeptide that inserts into the membrane in a calcium-dependent manner and depolarises it (bactericidal against MRSA and VRE, and classically inactivated by pulmonary surfactant so it is never used for pneumonia), and fusidic acid, which blocks elongation factor G. Drugs are also classed as bactericidal or bacteriostatic, and as concentration-dependent (aminoglycosides/fluoroquinolones) or time-dependent (beta-lactams/glycopeptides) killers - which determines dosing.
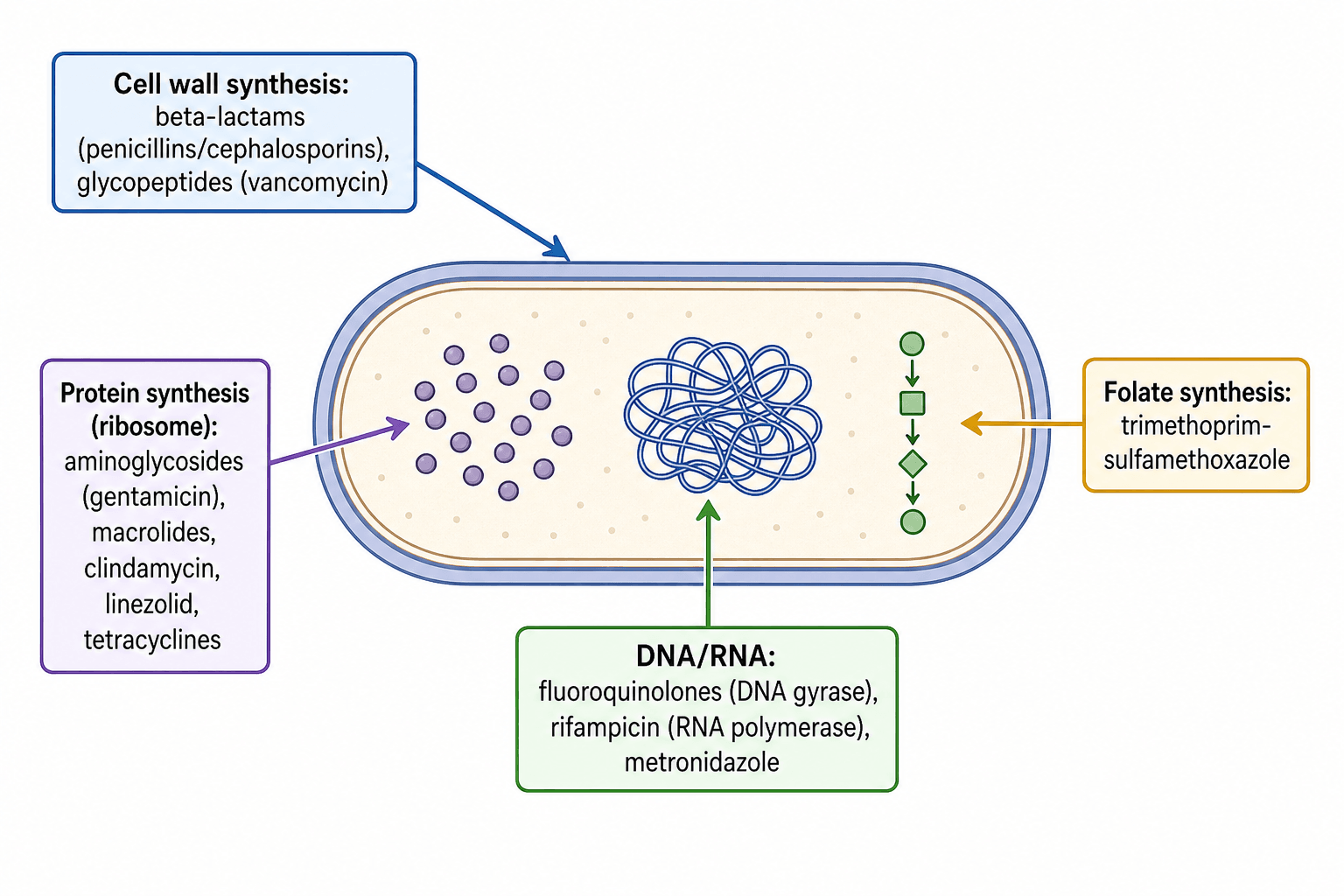
Resistance Mechanisms
Bacteria resist antibiotics by four mechanisms:
- Enzymatic inactivation: beta-lactamases (penicillinases, extended-spectrum beta-lactamases/ESBL, carbapenemases) and aminoglycoside-modifying enzymes destroy the drug.
- Target modification: an altered penicillin-binding protein (PBP2a, encoded by mecA) in MRSA; altered cell-wall precursor (D-ala-D-lac via vanA) in VRE; mutated DNA gyrase (fluoroquinolone resistance) or ribosome.
- Reduced uptake / decreased permeability: loss of outer-membrane porins (gram-negatives).
- Efflux pumps: actively pump the drug out. Resistance genes spread by plasmids/transposons (horizontal gene transfer), producing MRSA, VRE, ESBL and multidrug-resistant gram-negatives - relevant to surgical prophylaxis and the empirical treatment of infection.
Biofilm: The Orthopaedic Key
A biofilm is a structured community of bacteria adherent to a surface (implant, dead bone/sequestrum) within a self-produced extracellular matrix (glycocalyx). It causes antibiotic TOLERANCE - a phenotypic, reversible survival distinct from genetic resistance - through reduced antibiotic penetration into the matrix, slow-growing/dormant PERSISTER cells and SMALL-COLONY VARIANTS that survive antibiotic exposure, and an altered local microenvironment. The concept is quantified by the MBEC (minimum biofilm eradication concentration), which may be hundreds to over a thousand times the MIC of the same organism grown planktonically - so a laboratory report of "sensitive", derived from a planktonic MIC, says almost nothing about whether the drug will clear a biofilm. This is why antibiotics alone fail to clear implant-associated infection and why SURGICAL source control - debridement and often implant removal - is essential, combined with antibiotics. RIFAMPICIN penetrates the staphylococcal biofilm and intracellular bacteria, making it a key agent for staphylococcal implant infection, but it must always be given in COMBINATION (commonly with a fluoroquinolone) because rifampicin monotherapy rapidly selects resistance.
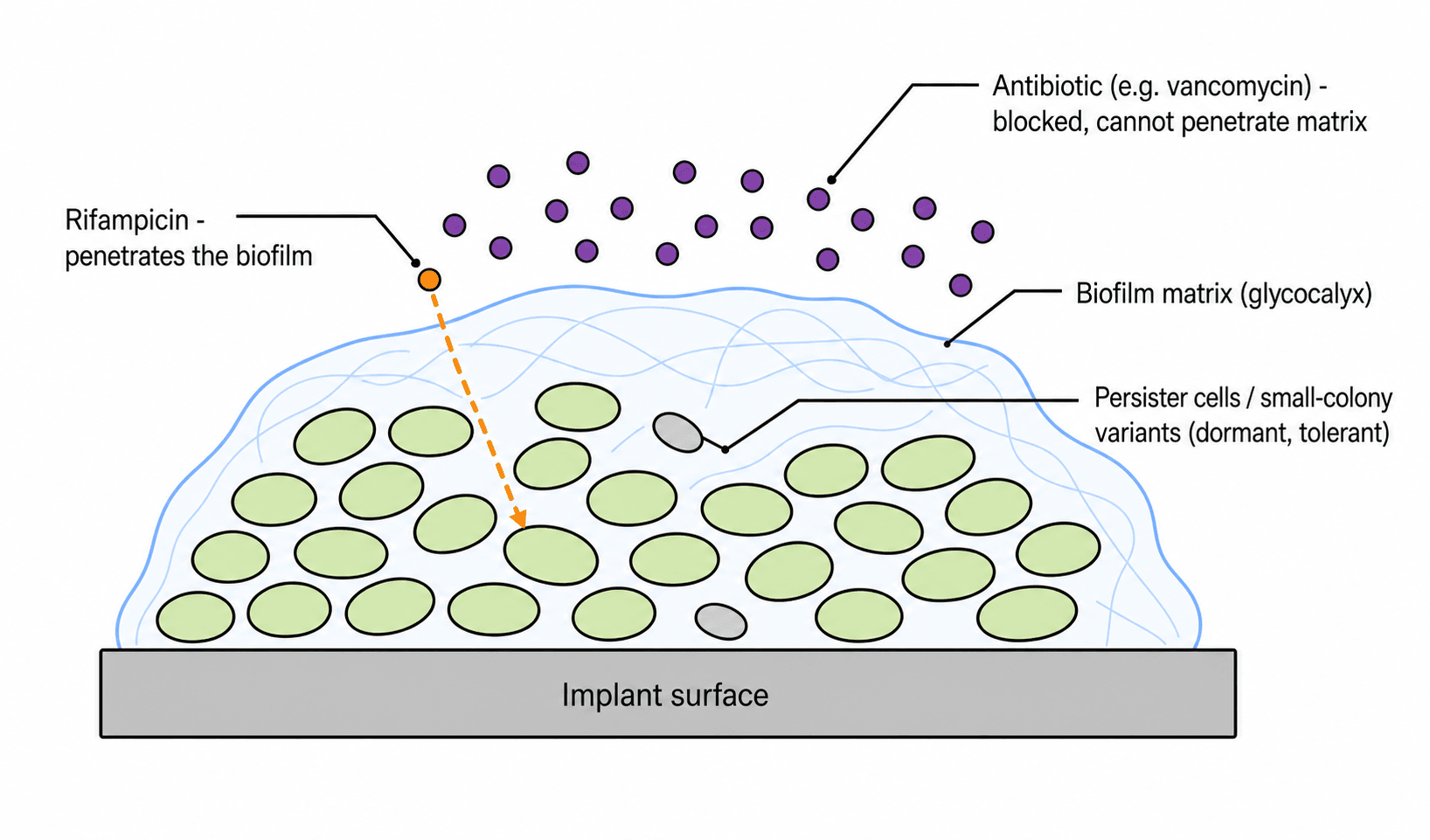
Two cardinal rules in implant infection: first, rifampicin is never used as monotherapy - resistance emerges rapidly, so it is always combined with another biofilm-relevant agent (e.g. a fluoroquinolone for susceptible organisms); and second, antibiotics cannot 'sterilise' a mature biofilm on a foreign body - durable cure needs surgical debridement and (usually) implant removal/exchange alongside antibiotics. Suppressive antibiotics may control, but rarely eradicate, biofilm infection when the implant is retained. Choose agents with good bone penetration (e.g. fluoroquinolones, clindamycin) and use local delivery (gentamicin/vancomycin cement/beads) as an adjunct.
Toxicities & Monitoring the Surgeon Must Know
These are the toxicities that change orthopaedic practice and require monitoring:
- Fluoroquinolones: tendinopathy and tendon rupture (classically the Achilles) - highly relevant to the orthopaedic patient - plus QT prolongation, dysglycaemia, C. difficile, and a cartilage/arthropathy caution in children (relative contraindication in the growing patient).
- Aminoglycosides (gentamicin): dose-related nephrotoxicity and ototoxicity (vestibular and cochlear) → therapeutic drug monitoring; relevant to systemic use and to high local cement/bead loads.
- Vancomycin (glycopeptide): nephrotoxicity and the rate-related "red man"/infusion reaction (not a true allergy) → level/AUC monitoring.
- Rifampicin: a potent hepatic enzyme inducer (drug interactions - reduces warfarin, oral contraceptives and many drugs), hepatotoxicity, and orange discoloration of secretions; and rapid resistance if used alone.
- Linezolid: with prolonged use, myelosuppression (thrombocytopenia), peripheral/optic neuropathy, and serotonin syndrome (it is a weak MAO inhibitor).
- Clindamycin: the classic association with C. difficile colitis.
- Beta-lactams: allergy/anaphylaxis (and a low penicillin-cephalosporin cross-reactivity).
The surgeon-facing high-yield ones are fluoroquinolone tendinopathy and aminoglycoside/vancomycin nephro-ototoxicity needing level monitoring.
Know the toxicities: fluoroquinolone tendinopathy/Achilles rupture (and paediatric cartilage caution); aminoglycoside nephro/ototoxicity and vancomycin nephrotoxicity/red-man (monitor levels); rifampicin enzyme induction/orange secretions; linezolid thrombocytopenia/serotonin syndrome; clindamycin C. difficile; beta-lactam allergy.
PK/PD & Bone Penetration: Operationalising the Dosing
-
The PK/PD indices: concentration-dependent killing is optimised by a high peak relative to the MIC (Cmax/MIC) and the AUC/MIC ratio - so aminoglycosides are given as a high once-daily dose (also exploiting the post-antibiotic effect), and fluoroquinolones are dosed to AUC/MIC. Time-dependent killing depends on the time the concentration stays above the MIC - so beta-lactams benefit from more frequent dosing or extended/continuous infusions, and vancomycin is dosed to an AUC/MIC target.
-
The definitions: the MIC (minimum inhibitory concentration) and MBC (minimum bactericidal concentration) underpin "bactericidal versus bacteriostatic".
-
Bone penetration (orthopaedically critical): fluoroquinolones, clindamycin, rifampicin, linezolid and fusidic acid penetrate bone well (useful oral options for osteomyelitis/PJI and oral switch), whereas beta-lactams and especially glycopeptides (vancomycin) penetrate bone relatively poorly - which informs agent and route choice and the appeal of bone-penetrating oral agents.
-
Oral bioavailability - why an oral switch is even possible. Several of the bone-penetrating agents are almost completely absorbed: fluoroquinolones, linezolid, metronidazole and fusidic acid approach near-complete oral bioavailability, and clindamycin and rifampicin are well absorbed too. For these drugs the oral route achieves serum concentrations close to intravenous, so an oral regimen is a pharmacological equivalent rather than a compromise. By contrast vancomycin is not absorbed orally at all - oral vancomycin treats C. difficile within the gut lumen and has no systemic activity whatever.
-
Half-life drives redosing. Intra-operative redosing is a pharmacokinetic decision, not a ritual: a further dose is due once the operation exceeds roughly two half-lives of the agent, or after major blood loss, because the aim is a tissue concentration above the MIC for the whole time the wound is open. Obesity raises the volume of distribution and is the usual reason a standard dose under-doses.
-
The monitoring target. Vancomycin is now dosed to an AUC/MIC of roughly 400-600, which has largely replaced trough-only monitoring because it tracks efficacy while limiting nephrotoxicity.
-
Synergy. Some combinations kill better than either agent alone - classically a beta-lactam (or vancomycin) plus an aminoglycoside in enterococcal infection, where cell-wall disruption lets the aminoglycoside reach its ribosomal target.
(Prophylaxis regimens, timing and the penicillin-allergy pathway are developed in our Perioperative Antibiotics topic; treatment regimens, duration and the oral-versus-intravenous evidence in Orthopaedic Antibiotic Therapy; cement and beads in Local Antibiotic Delivery. The pharmacology that drives all three sits here.)
Concentration-dependent (aminoglycosides, fluoroquinolones): high peak/MIC and AUC/MIC → high once-daily dosing (post-antibiotic effect); time-dependent (beta-lactams, vancomycin): time above the MIC → frequent/extended infusions. Bone penetration good for fluoroquinolones/clindamycin/rifampicin/ linezolid/fusidic acid, poor for vancomycin/beta-lactams.
Reading a Susceptibility Report, and Stewardship
The microbiology report is where pharmacology meets the ward round, and it is routinely over-interpreted.
- "Sensitive" is a planktonic statement. Breakpoints (S/I/R) are set against a planktonic MIC and a set of assumptions about achievable serum concentration at a standard dose. They say nothing about whether the drug reaches bone, and nothing about a biofilm on an implant - which is precisely why an organism reported sensitive to vancomycin can persist on a prosthesis. Read the report alongside the site of infection and whether a foreign body is present.
- "Intermediate" is usually a dosing statement, not a prohibition: it generally means the agent may work at a higher dose or where the drug concentrates.
- Sampling before antibiotics. Antibiotics given before deep samples are taken substantially reduce culture yield and are a common reason for a culture-negative bone or joint infection. Unless the patient is septic, hold antibiotics until adequate deep samples - usually multiple - have been obtained.
- Selective pressure and collateral damage. Every course selects resistance in the patient's own flora. Clindamycin, fluoroquinolones and broad-spectrum cephalosporins carry the strongest association with C. difficile; broad gram-negative cover selects ESBL and resistant Pseudomonas. Choosing the narrowest effective agent is a clinical decision, not an administrative one.
- Stewardship in orthopaedic terms means: prophylaxis is a single perioperative course, not a post-operative drip continued "until the drains are out"; empirical therapy is de-escalated once cultures return; duration is defined at the outset rather than open-ended; and the oral switch is taken as soon as the agent's bioavailability and bone penetration allow.
Mnemonics & Memory Aids
WRDF (targets)
Hook:Antibiotic targets = Wall, Ribosome, DNA/RNA, Folate (WRDF).
BIOFILM
Hook:BIOFILM = tolerance; treat with surgery + biofilm-active antibiotics.
Clinical Decision Scenarios
Practise clinical reasoning and management decisions out loud
“Classify antibiotics by mechanism, and explain the main mechanisms of bacterial resistance.”
“What is biofilm, and why does it mean an infected implant usually cannot be cured by antibiotics alone?”
Classes by target
- Cell wall: beta-lactams (PBPs), glycopeptides (vancomycin - D-ala-D-ala)
- Ribosome: aminoglycosides, macrolides, clindamycin, linezolid, tetracyclines
- DNA/RNA: fluoroquinolones (gyrase), rifampicin (RNA pol), metronidazole; folate: TMP-SMX
Killing
- Bactericidal (beta-lactams, glycopeptides, aminoglycosides, fluoroquinolones, rifampicin) vs bacteriostatic
- Concentration-dependent (aminoglycosides, fluoroquinolones)
- Time-dependent (beta-lactams, glycopeptides)
Resistance
- Enzymatic (beta-lactamases/ESBL/carbapenemases)
- Target modification (MRSA mecA/PBP2a; VRE vanA; gyrase mutation)
- Reduced uptake (porin loss); efflux pumps; spread by plasmids
Biofilm (orthopaedic key)
- Matrix on implant -> tolerance (reduced penetration, persisters, small-colony variants)
- Antibiotics alone FAIL -> surgical debridement/implant removal + antibiotics
- Rifampicin penetrates staph biofilm (ALWAYS combination); use bone-penetrating agents + local delivery
Evidence & Key Studies
Biofilm-mediated antibiotic tolerance precedes resistance in Staphylococcus aureus under antibiotic selection
- In periprosthetic joint infection, bacteria survive antibiotics through biofilm-mediated TOLERANCE, resistance and persistence - antibiotics in isolation are typically ineffective.
- Rifampicin penetrates the S. aureus biofilm whereas vancomycin penetrates poorly; tolerant small-colony variants emerge under antibiotic selective pressure.
- Biofilm-mediated tolerance explains why treatments relying on antibiotics to clear residual biofilm fail - reinforcing the need for surgical source control.
Antimicrobial efficacy differs between planktonic and biofilm bacterial phenotypes (levofloxacin, rifampin)
- Antibiotics have markedly different efficacy against bacteria in the planktonic versus the biofilm phenotype (higher minimum bactericidal concentrations against biofilm).
- Levofloxacin and rifampin were studied against S. aureus biofilm on an implant model, underscoring biofilm tolerance.
- The bacterial phenotype (biofilm) is a key determinant of treatment efficacy for implant infections.
The central role of biofilm-mediated tolerance (with persister cells/small-colony variants) in periprosthetic joint infection, and rifampicin's biofilm penetration versus vancomycin's poor penetration, come from the cited Manasherob study, and the planktonic-versus-biofilm efficacy difference from the cited Ong study. The antibiotic-class mechanisms, bactericidal/bacteriostatic and concentration-/time- dependent killing, and the four resistance mechanisms (including MRSA mecA and VRE vanA) are standard, well-established pharmacology. (See also our Periprosthetic Joint Infection, Osteomyelitis, Local Antibiotic Delivery and Septic Arthritis topics.)