Multiple Enchondromas | IDH Mutations | High Malignancy Risk | Lifelong Surveillance Required
- Ollier disease - multiple unilateral enchondromas, 25-30% malignant transformation by age 40
- Maffucci syndrome - enchondromas plus hemangiomas, nearly 100% lifetime malignancy risk
- IDH1 and IDH2 somatic mutations found in 87% of enchondromas and secondary chondrosarcomas
- Lifelong surveillance required with annual clinical exam and imaging of symptomatic lesions
- Pain, growth, or soft tissue mass in any lesion requires urgent MRI and biopsy consideration
- “Ollier disease is sporadic (somatic IDH mutations), NOT inherited - low recurrence risk
- “Maffucci syndrome patients develop both chondrosarcoma (from enchondromas) and angiosarcoma (from hemangiomas)
- “Deformities common: limb length discrepancy, angular deformity, pathological fractures
- “Low threshold for biopsy - any changing lesion is malignant until proven otherwise
Ollier disease carries 25-30% risk of chondrosarcoma by age 40. Maffucci syndrome has nearly 100% lifetime malignancy risk - both chondrosarcoma from enchondromas and angiosarcoma from hemangiomas. Any new pain, growth, or soft tissue mass requires urgent MRI and biopsy.
Ollier disease shows unilateral or predominantly one-sided distribution of enchondromas. If bilateral and symmetric, consider Maffucci syndrome (if hemangiomas present) or metachondromatosis (if osteochondromas present).
IDH1 and IDH2 somatic mutations found in 87% of enchondromas and chondrosarcomas in Ollier/Maffucci. These are NOT germline mutations - disease is sporadic, not inherited. Mutations disrupt cartilage differentiation leading to multiple cartilage rests.
Annual clinical examination with skeletal survey. MRI any symptomatic lesion - pain, growth, or palpable mass. Low threshold for biopsy of suspicious lesions. Early detection of malignant transformation is critical for limb salvage and survival.
- Solitary Enchondroma
- Single lesion
- Ollier Disease
- Multiple (5 or more typical)
- Maffucci Syndrome
- Multiple plus hemangiomas
- Solitary Enchondroma
- Usually hand/foot
- Ollier Disease
- Unilateral or asymmetric
- Maffucci Syndrome
- Asymmetric, any bone
- Solitary Enchondroma
- 1-2% lifetime
- Ollier Disease
- 25-30% by age 40
- Maffucci Syndrome
- Nearly 100% lifetime
- Solitary Enchondroma
- Sporadic, usually no mutation
- Ollier Disease
- Somatic IDH1/IDH2 (87%)
- Maffucci Syndrome
- Somatic IDH1/IDH2 (87%)
- Solitary Enchondroma
- None required if asymptomatic
- Ollier Disease
- Annual exam and imaging
- Maffucci Syndrome
- Aggressive annual surveillance
- Solitary Enchondroma
- Rare
- Ollier Disease
- Common (LLD, angulation)
- Maffucci Syndrome
- Common plus vascular lesions
Overview and Epidemiology
Enchondromatosis refers to syndromes characterized by multiple enchondromas. The two main types are:
Ollier disease: Multiple enchondromas with unilateral or asymmetric distribution, NO soft tissue hemangiomas.
Maffucci syndrome: Multiple enchondromas PLUS soft tissue hemangiomas (spindle cell hemangiomas).
Both are sporadic conditions caused by somatic mutations in IDH1 or IDH2 genes, NOT inherited. Recurrence risk in offspring is very low.
- Incidence: Approximately 1 in 100,000 live births
- Age at presentation: Childhood (under 10 years typical)
- Sex distribution: Equal male to female
- Distribution: Unilateral or markedly asymmetric
- Malignancy risk: 25-30% by age 40 years
- Incidence: Even rarer than Ollier (1 in 1,000,000)
- Age at presentation: Childhood, often younger than Ollier
- Hemangiomas: Spindle cell type, visible as soft tissue masses
- Malignancy risk: Nearly 100% lifetime risk
- Tumor types: Chondrosarcoma AND angiosarcoma
Historical Context
The term Ollier disease was named after French surgeon Louis Ollier who described multiple enchondromatosis in 1899. Maffucci syndrome was described by Italian pathologist Angelo Maffucci in 1881 who recognized the association between enchondromas and hemangiomas.
Historically, these conditions were poorly understood and outcomes were dismal. Modern understanding of IDH mutations (discovered 2011) and improved surveillance protocols have enabled earlier detection of malignant transformation and improved survival.
Pathophysiology
IDH1 and IDH2 Mutations
In 2011, Pansuriya et al discovered that 87% of enchondromas and secondary chondrosarcomas in Ollier disease and Maffucci syndrome harbor somatic mutations in IDH1 (most common) or IDH2 genes. These are:
- Somatic mutations (NOT germline) - explains sporadic occurrence
- Mosaic distribution - mutations occur early in development affecting specific tissue populations
- Present in BOTH benign and malignant lesions - suggests additional genetic hits required for malignant transformation
- Same mutations seen in gliomas - explains increased brain tumor risk in Maffucci syndrome
Key point: IDH mutations are present in enchondromas from birth but malignant transformation requires additional genetic changes (TP53, RB1 mutations) that accumulate over time.
Pathogenesis of Enchondromatosis
Somatic IDH mutation occurs in mesenchymal stem cell during early development. Mutation causes abnormal accumulation of 2-hydroxyglutarate (2-HG) which disrupts cartilage differentiation and normal enchondral ossification.
Multiple cartilage rests persist in medullary cavity instead of ossifying. These appear as enchondromas on X-ray. Growth plate abnormalities lead to limb length discrepancy and angular deformities as child grows.
Accumulation of additional mutations (TP53, RB1, CDKN2A) in some lesions drives malignant transformation. Pain, growth, cortical breakthrough, and soft tissue mass indicate transformation to chondrosarcoma.
For Maffucci syndrome patients, hemangiomas may also transform to angiosarcoma. Multiple tumor sites possible. Prognosis poor due to multifocal disease and late presentation.
The 2-Hydroxyglutarate Oncometabolite — How the IDH Mutation Actually Drives Disease
The topic repeatedly invokes 2-hydroxyglutarate (2-HG), "epigenetic dysregulation", "DNA hypermethylation" and the mutant-IDH1 drug ivosidenib, so it is worth joining these together into the single mechanism that underlies the whole disease.
- Normal IDH1/IDH2 convert isocitrate to alpha-ketoglutarate (alpha-KG) in cellular metabolism.
- The disease-causing mutations (IDH1 R132, IDH2 R172) are neomorphic gain-of-function changes: the mutant enzyme does not simply lose function - it acquires a new activity, reducing alpha-KG to the oncometabolite D-2-hydroxyglutarate (D-2-HG), which accumulates to very high levels.
- D-2-HG closely resembles alpha-KG and therefore competitively inhibits alpha-KG-dependent dioxygenases, most importantly the TET DNA demethylases and the Jumonji-domain histone demethylases (and prolyl-hydroxylases that regulate HIF).
- Inhibiting these demethylases produces DNA and histone hypermethylation (exactly the hypermethylation Pansuriya found), which silences differentiation genes and arrests mesenchymal/chondrocyte maturation - so cartilage rests persist as enchondromas instead of undergoing normal endochondral ossification.
- IDH mutation alone is insufficient for malignancy (it is present in both benign and malignant lesions), which is why transformation needs the additional hits (CDKN2A, TP53, RB1) described above.
- Therapeutic and biomarker relevance: because the lesions are built on 2-HG, mutant-IDH1 inhibition with ivosidenib lowers plasma 2-HG toward normal and stabilises some advanced IDH-mutant chondrosarcomas (see the evidence base), and circulating 2-HG can serve as a disease biomarker.
The IDH mutation in Ollier/Maffucci is neomorphic, not merely loss-of-function: the mutant enzyme gains the ability to make the oncometabolite D-2-hydroxyglutarate, an alpha-ketoglutarate mimic that competitively inhibits TET DNA demethylases and Jumonji histone demethylases, causing hypermethylation that blocks chondrocyte differentiation. This single mechanism explains the sporadic mosaic enchondromas, the shared mutation in the Maffucci haemangiomas, and why ivosidenib (a mutant-IDH1 inhibitor that lowers 2-HG) is being trialled in IDH-mutant chondrosarcoma.
Histopathology
- Cellularity: Hypocellular hyaline cartilage
- Chondrocytes: Small uniform cells in lacunae
- Nuclei: Single, regular, small nuclei
- Matrix: Abundant hyaline cartilage matrix
- Calcification: Dystrophic calcification common
- Growth pattern: Lobular architecture preserved
- Cellularity: Hypercellular (increased cell density)
- Chondrocytes: Enlarged cells, irregular distribution
- Nuclei: Enlarged hyperchromatic nuclei, binucleation
- Matrix: Myxoid degeneration areas
- Permeation: Infiltration into surrounding bone
- Atypia: Moderate to severe nuclear atypia
Distinguishing benign from malignant lesions histologically is extremely difficult in enchondromatosis patients. Key challenges:
- Enchondromas in Ollier/Maffucci may show higher cellularity than solitary enchondromas (still benign)
- Low-grade chondrosarcoma may have minimal atypia (subtle changes)
- Sampling error common - biopsy may miss malignant areas in heterogeneous tumors
Clinical and radiological correlation is ESSENTIAL. Pain, growth, cortical destruction, and soft tissue mass are more reliable indicators of malignancy than histology alone. Expert musculoskeletal pathologist review mandatory.
Classification
Types of Enchondromatosis
- Key Features
- Multiple enchondromas, predominantly unilateral distribution, limb shortening
- Extra-Skeletal Findings
- None
- Malignancy Risk
- 25-30% lifetime risk of chondrosarcoma
- Key Features
- Multiple enchondromas + multiple soft tissue hemangiomas
- Extra-Skeletal Findings
- Spindle cell hemangiomas (pathognomonic)
- Malignancy Risk
- Higher risk (40-50%), plus risk of other malignancies
- Key Features
- Enchondromas + osteochondromas (exostoses)
- Extra-Skeletal Findings
- None
- Malignancy Risk
- Low
- Key Features
- Generalized enchondromatosis, autosomal dominant
- Extra-Skeletal Findings
- None
- Malignancy Risk
- Unknown
Distribution Patterns
- Typically unilateral or predominantly affects one side
- Commonly affects hands and feet
- Long bones of lower limb frequently involved
- May cause significant limb length discrepancy
- Similar enchondroma distribution to Ollier
- Plus spindle cell hemangiomas (soft tissue masses)
- Hemangiomas may be in soft tissues or viscera
- Higher overall malignancy risk
Exam Viva Point: "How do you differentiate Ollier disease from Maffucci syndrome?" Answer: Maffucci syndrome = enchondromas + soft tissue hemangiomas (spindle cell type). Both have IDH mutations as underlying cause. Maffucci has higher malignancy risk (40-50% vs 25-30%) and increased risk of non-skeletal malignancies (gliomas, ovarian tumors).
Classification is based on presence or absence of extra-skeletal features, particularly soft tissue hemangiomas.
Clinical Presentation
Typical Presentation Patterns
- Limb deformity noticed by parents (leg length difference)
- Palpable masses in hands or feet (cartilage enlargement)
- Gait abnormality due to limb length discrepancy
- Incidental X-ray finding for minor trauma
- Angular deformity (varus/valgus) of long bones
- Pathological fracture through weakened bone
- Progressive deformity worsening with growth
- Pain in lesion (RED FLAG - malignant transformation)
- Palpable soft tissue mass (chondrosarcoma with soft tissue extension)
- Functional impairment from severe deformities
Skeletal Deformities
- Mechanism
- Asymmetric growth plate involvement
- Location
- Lower limbs most common
- Management
- Epiphysiodesis or lengthening
- Mechanism
- Metaphyseal enchondromas disrupt growth
- Location
- Tibia, femur, forearm
- Management
- Corrective osteotomy when severe
- Mechanism
- Multiple phalangeal enchondromas
- Location
- Fingers shortened and widened
- Management
- Curettage after fractures only
- Mechanism
- Cortical thinning from lesions
- Location
- Any involved bone
- Management
- Stabilize, then curettage after healing
Red Flag Symptoms - Malignant Transformation
Any of these symptoms in a known enchondromatosis patient requires URGENT MRI and biopsy consideration:
- New onset pain in previously asymptomatic lesion (MOST IMPORTANT)
- Progressive enlargement on serial X-rays
- Palpable soft tissue mass on examination
- Cortical breakthrough on X-ray or CT
- Rapid functional decline (new weakness, limited motion)
- Night pain or pain at rest
- Constitutional symptoms (rare - weight loss, fatigue)
Management: MRI of affected area, CT chest to rule out metastases, CT-guided biopsy with excisable trajectory, multidisciplinary tumor board discussion. Do NOT delay - early detection critical for limb salvage.
Physical Examination
Systematic Examination of Enchondromatosis Patient
- Limb lengths: Measure leg lengths (ASIS to medial malleolus)
- Angular deformities: Assess varus/valgus alignment of limbs
- Hand deformities: Note shortened digits, expanded phalanges
- Soft tissue masses: Look for hemangiomas (Maffucci) or soft tissue extension of tumors
- Gait: Observe for limp, Trendelenburg gait
- Bony masses: Palpate all visible/palpable lesions
- Tenderness: Any tender lesion is RED FLAG for malignancy
- Soft tissue masses: Palpate for extraosseous extension
- Hemangiomas: Compressible soft tissue masses (Maffucci)
- Temperature: Warm areas suggest active tumor growth
- Joint range of motion: Check all major joints
- Limb rotation: Assess for rotational deformities
- Functional assessment: Grip strength, walking distance
- Neurovascular exam: Check pulses, sensation in all limbs
- Skeletal diagram: Map all known lesions
- Photography: Document visible deformities and hemangiomas
- Measurements: Record limb lengths and joint angles
- Pain assessment: Document any painful lesions for urgent MRI
Investigations and Imaging
Baseline Skeletal Survey
At diagnosis, patients require complete skeletal survey to document all lesions and establish baseline for surveillance.
Initial Imaging Protocol
- All extremities: AP and lateral views
- Pelvis and femurs: AP views
- Spine: AP and lateral if symptomatic
- Hands and feet: PA views
- Document: number of lesions, size, cortical involvement, deformities
- Proximal long bones: Femur, humerus (high malignancy risk sites)
- Painful lesions: Any lesion with new pain
- Large lesions: Greater than 5cm or significant cortical thinning
- Assess: soft tissue extension, marrow involvement, cortical integrity
- Clinical photos: All visible deformities
- Limb length measurements: Scanogram or CT scanogram
- Angular measurements: Long-leg alignment films if needed
Radiographic Features of Enchondromas (Ollier/Maffucci)
- Appearance
- Rings-and-arcs or stippled pattern
- Location
- Within medullary cavity
- Significance
- Pathognomonic for cartilage tumor
- Appearance
- Endosteal scalloping, expansion
- Location
- Circumferential in severe cases
- Significance
- Pathological fracture risk
- Appearance
- Metaphyseal location, crosses physis
- Location
- Long bones near joints
- Significance
- Causes growth disturbance and deformity
- Appearance
- Multiple lesions, unilateral predominance
- Location
- Entire limb or hemibody
- Significance
- Diagnostic for Ollier disease
Phleboliths and the Spindle Cell Haemangioma — The Maffucci Imaging Hallmark
The radiographic features above describe the enchondromas, but in Maffucci syndrome the diagnosis is frequently made on the plain film by spotting the soft-tissue vascular lesions alongside them - a point the topic relies on (phleboliths and "spindle cell haemangiomas (pathognomonic)") but never develops.
- Phleboliths: small round or ovoid calcified thrombi (often laminated, target-like, sometimes with a lucent centre) that form in the slow-flowing channels of the soft-tissue haemangiomas. Multiple soft-tissue phleboliths next to multiple enchondromas in a young patient are effectively pathognomonic of Maffucci syndrome and are the single most useful plain-film clue that separates it from Ollier disease, which has no soft-tissue vascular calcification.
- The spindle cell haemangioma: the "haemangioma" of Maffucci is specifically a spindle cell haemangioma, now regarded as a benign vascular neoplasm (rather than a true haemangioma or a simple malformation), typically in the dermis/subcutis of the distal extremities, composed of cavernous vascular spaces (with thrombi/phleboliths) and bland spindled cells. Crucially it carries the same somatic IDH1/IDH2 mutation as the enchondromas (found in about 70 percent of spindle cell haemangiomas), molecular proof that the two Maffucci lesions share an origin.
- Cross-sectional imaging: the vascular lesions appear as soft-tissue masses containing phleboliths on radiograph/CT and as lobulated, markedly T2-hyperintense vascular spaces (sometimes with fluid-fluid levels) on MRI. The key surveillance concern is transformation to angiosarcoma (rapid growth, new firmness or pain), managed as described later.
In a child or young adult with multiple enchondromas, look at the soft tissues for phleboliths - round calcified densities outside bone. Their presence signals the spindle cell haemangiomas of Maffucci syndrome (with its far higher, dual chondrosarcoma-plus-angiosarcoma malignancy risk and the same IDH mutation), whereas their absence supports Ollier disease. It is the quickest plain-film discriminator between the two.
MRI Assessment for Malignancy
MRI is the gold standard for detecting malignant transformation. Key features:
- T1 signal: Low to intermediate (cartilage)
- T2 signal: Very high (hyaline cartilage water content)
- Enhancement: Peripheral septal enhancement only
- Soft tissue: No extraosseous component
- Margins: Well-defined lobulated contour
- Size: Variable but stable on serial imaging
- T1 signal: Heterogeneous with low signal areas
- T2 signal: Heterogeneous (myxoid areas)
- Enhancement: Intense irregular enhancement
- Soft tissue: Soft tissue mass PRESENT
- Margins: Ill-defined, infiltrative pattern
- Size: Progressive enlargement on serial MRI
Presence of soft tissue mass on MRI is the SINGLE MOST RELIABLE indicator of malignant transformation in enchondromatosis patients. Benign enchondromas do NOT break through cortex and extend into soft tissue. If you see soft tissue component, assume chondrosarcoma until proven otherwise and proceed with biopsy and wide excision.
Imaging Atlas


Surveillance Imaging Protocol
- Clinical Exam
- Annual full exam, measure limbs
- Imaging
- Skeletal survey every 2-3 years
- Frequency
- More frequent if growing deformities
- Clinical Exam
- Annual exam, document new symptoms
- Imaging
- X-rays of symptomatic areas only
- Frequency
- Annual clinical, imaging as needed
- Clinical Exam
- Annual exam with high suspicion
- Imaging
- Low threshold for MRI if any symptoms
- Frequency
- Annual, more aggressive imaging
- Clinical Exam
- Urgent clinical evaluation
- Imaging
- MRI of affected area, CT chest
- Frequency
- Immediate workup for malignancy
Biopsy Decision-Making
When to Biopsy in Enchondromatosis
Perform biopsy if:
- New pain in previously asymptomatic lesion
- Progressive enlargement on serial imaging
- Soft tissue mass on MRI
- Cortical breakthrough on CT
- Patient age over 40 with new symptoms in axial skeleton
Technique: CT-guided core needle biopsy with excisable trajectory (plan for wide excision through same approach if malignant).
Discuss at tumor board:
- Large lesion (over 5cm) in proximal long bone
- Moderate cortical destruction (over 2/3 thickness)
- Heterogeneous enhancement on MRI (equivocal)
- Patient anxiety about specific lesion
Balance risk of tumor seeding against diagnostic benefit.
Safe to observe:
- Small asymptomatic hand/foot lesions
- Stable size on serial imaging (2+ years)
- Classic benign MRI features if MRI performed
- Young patient with no concerning features
Serial imaging every 6-12 months safer than biopsy.
Specific considerations for biopsy in Ollier/Maffucci patients:
- Tumor seeding: Risk of seeding biopsy tract with malignant cells
- Sampling error: Heterogeneous tumors may show benign areas in biopsy but malignant areas elsewhere
- Histological overlap: Even expert pathologists struggle to distinguish benign from low-grade malignant
- Multiple lesions: Difficult to biopsy every concerning lesion
Recommendation: Reserve biopsy for lesions where clinical/imaging strongly suggests malignancy AND where result will change management (i.e., proceed with wide excision if confirmed chondrosarcoma).
Differential Diagnosis
- Cartilage Lesions
- Multiple enchondromas, unilateral
- Other Features
- Deformities, NO hemangiomas
- Malignancy Risk
- 25-30% chondrosarcoma
- Cartilage Lesions
- Multiple enchondromas
- Other Features
- Soft tissue hemangiomas present
- Malignancy Risk
- Nearly 100% (chondrosarcoma + angiosarcoma)
- Cartilage Lesions
- Enchondromas + osteochondromas
- Other Features
- Autosomal dominant, PTPN11 mutation
- Malignancy Risk
- Low malignancy risk
- Cartilage Lesions
- Osteochondromas only (NO enchondromas)
- Other Features
- Autosomal dominant, EXT1/EXT2
- Malignancy Risk
- 1-5% malignant transformation
Ollier vs Maffucci: Look for soft tissue hemangiomas (visible, compressible masses). If present, it's Maffucci with nearly 100% malignancy risk. If absent, it's Ollier with 25-30% risk.
Enchondromas vs Osteochondromas: Enchondromas are INTRAMEDULLARY (inside bone) with rings-and-arcs calcification. Osteochondromas are SURFACE lesions (exostoses) with cartilage cap pointing away from joint. Metachondromatosis has BOTH types.
Ollier vs Metachondromatosis: Metachondromatosis is autosomal dominant (family history), has both enchondromas and osteochondromas, and has MUCH lower malignancy risk than Ollier.
Management Algorithm
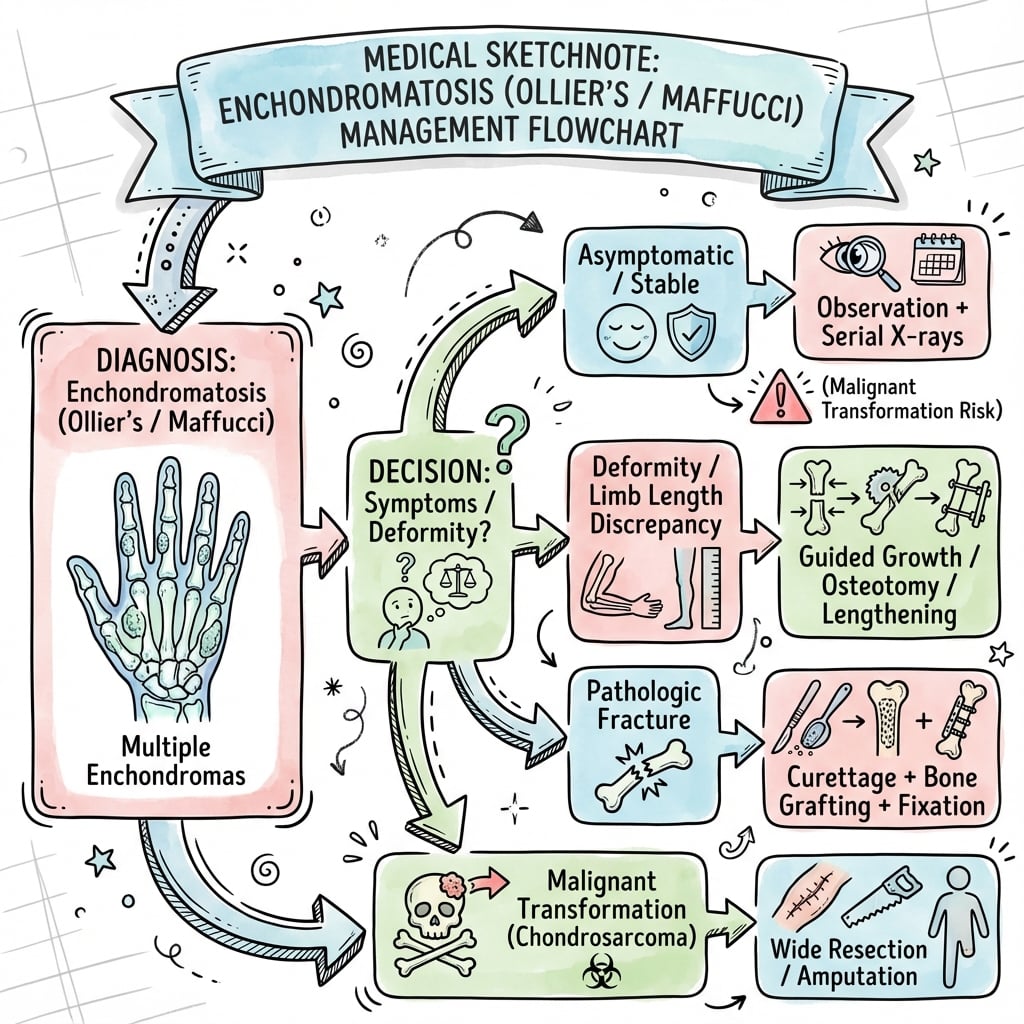
Lifelong Surveillance Protocol
Goal: Early detection of malignant transformation to enable limb-salvage surgery and improve survival.
Annual Surveillance Visit
- Pain assessment: Any new pain in any lesion? Character, duration, severity
- Functional change: New weakness, limited motion, gait change?
- Visible changes: New masses, enlarging lesions?
- Constitutional symptoms: Weight loss, fatigue, night sweats?
- Fractures: Any new pathological fractures since last visit?
- Inspect all limbs: Look for new deformities, masses, asymmetry
- Palpate all accessible lesions: Check for tenderness (RED FLAG)
- Measure limb lengths: Document progression of discrepancy
- Assess deformities: Angular deformities, rotational abnormalities
- Check hemangiomas: In Maffucci, assess for changes (angiosarcoma)
- X-rays: Any symptomatic areas or areas of concern on exam
- MRI: LOW threshold - any painful lesion or palpable change
- Comparison: Compare to previous imaging for subtle growth
- CT chest: If chondrosarcoma suspected (staging)
- Warning signs: Teach patient red flags (pain, swelling, growth)
- Self-examination: How to palpate accessible lesions monthly
- Urgent reporting: Instruct to report new symptoms immediately
- Prognosis: Honest discussion of malignancy risk
Early detection of malignant transformation is CRITICAL. Studies show that chondrosarcomas detected early (small size, no metastases) have 90% 5-year survival with wide excision. Late detection (large size, metastatic) has under 30% survival. Aggressive surveillance and low threshold for MRI/biopsy are essential.
Surveillance Imaging Schedule
- Baseline Imaging
- Skeletal survey at diagnosis
- Follow-up Imaging
- Repeat skeletal survey every 2-3 years
- Indications
- Monitor growth, detect new lesions
- Baseline Imaging
- Update skeletal survey if not recent
- Follow-up Imaging
- X-rays of symptomatic areas annually
- Indications
- Peak age for malignant transformation
- Baseline Imaging
- Skeletal survey if new symptoms
- Follow-up Imaging
- MRI any new pain or growth immediately
- Indications
- Very high suspicion for malignancy
This completes the surveillance section.
Surgical Technique
Surgical Indications
- Pathological fracture (after healing for curettage)
- Symptomatic lesions causing pain or dysfunction
- Angular deformity affecting function
- Limb length discrepancy greater than 2cm
- Suspected malignant transformation
- Cosmetically unacceptable deformity
- Asymptomatic lesions (surveillance preferred)
- Active malignancy without staging workup
- Poor soft tissue envelope
- Multiple procedures planned (stage appropriately)
- Growing skeleton (consider timing)
Curettage and Bone Grafting
- Technique
- Adequate cortical window for complete visualization
- Key Points
- Window 2/3 length of lesion, hinge on one side for closure
- Technique
- Systematic removal of all cartilaginous tissue
- Key Points
- Start centrally, work to periphery; curettes of various sizes
- Technique
- High-speed burr to remove residual tissue
- Key Points
- Extends margins 1-2mm into normal bone
- Technique
- Phenol, hydrogen peroxide, or cryotherapy
- Key Points
- For aggressive lesions or recurrences; protect soft tissues
- Technique
- Fill defect with autograft, allograft, or substitute
- Key Points
- Autograft preferred for large defects; calcium phosphate for small lesions
- Technique
- Internal fixation if fracture risk high
- Key Points
- Prophylactic plating for large defects in weight-bearing bones
Hand Enchondroma Surgery
- Incision: Dorsal longitudinal or mid-lateral
- Window: Rectangular cortical window (preserve for closure)
- Curettage: Complete removal with small curettes
- Grafting: Often not required for small defects (fibrous healing)
- Fixation: K-wire only if unstable fracture
- Early motion: Begin at 2-4 weeks
- Multiple lesions: Stage surgeries 6-8 weeks apart
- Pathological fracture: Allow healing (4-6 weeks) before curettage
- Digital nerve: Protect during exposure
- Tendon adherence: Meticulous soft tissue handling
- No adjuvant: Rarely needed in hand
Deformity Correction
- Procedure
- Guided growth with 8-plate if growing
- Timing Considerations
- Remove at skeletal maturity or correction
- Procedure
- Acute corrective osteotomy
- Timing Considerations
- Wait until near skeletal maturity if possible
- Procedure
- Gradual correction with external fixator
- Timing Considerations
- Taylor Spatial Frame or Ilizarov
- Procedure
- Osteotomy with lengthening
- Timing Considerations
- Address both simultaneously with circular fixator
Limb Length Discrepancy Management
- Indication: Predicted discrepancy 2-5cm at maturity
- Timing: Based on growth remaining (Paley multiplier)
- Technique: Percutaneous drill or 8-plate
- Advantage: Simple, outpatient procedure
- Limitation: Cannot correct existing discrepancy
- Indication: Discrepancy greater than 5cm or skeletal maturity
- Rate: 1mm/day (0.25mm x 4 increments)
- External fixator: Ilizarov or TSF
- Internal lengthening nail: PRECICE, FITBONE
- Consolidation index: ~36 days/cm
Any lesion with pain at rest, rapid growth, cortical destruction, or soft tissue mass requires biopsy BEFORE definitive surgery. Suspected chondrosarcoma needs wide resection, NOT curettage.
Complications and Outcomes
Disease-Related Complications
- Incidence
- 25-30% Ollier, 100% Maffucci
- Impact
- Life-threatening, requires wide excision
- Management
- Surveillance, early detection, wide resection
- Incidence
- 30-40% of patients
- Impact
- Pain, disability, may require surgery
- Management
- Immobilize, heal, then curettage and graft
- Incidence
- 60-70% with lower limb involvement
- Impact
- Gait abnormality, back pain, cosmetic
- Management
- Epiphysiodesis or lengthening
- Incidence
- 40-50% with metaphyseal lesions
- Impact
- Joint malalignment, arthritis risk
- Management
- Guided growth or corrective osteotomy
- Incidence
- Variable (20-80%)
- Impact
- Limited activities, reduced quality of life
- Management
- Physiotherapy, adaptive equipment, surgery
Treatment-Related Complications
- Recurrence: 5-15% after curettage (incomplete removal)
- Re-fracture: 5-10% if large defect not adequately grafted
- Infection: 2-3% (standard surgical site infection risk)
- Stiffness: 10-20% in hand surgery without early mobilization
- Allograft nonunion: 10-20% at host-graft junction
- Allograft fracture: 5-10% years after surgery
- Prosthetic loosening: 5-10% at 10 years for endoprosthesis
- Infection: 5-15% for major reconstructions
- Limb length discrepancy: May persist after reconstruction
Long-Term Outcomes
Enchondromatosis patients face lifelong challenges:
Physical: Multiple surgeries (fractures, deformities, malignancy), chronic pain, functional limitations, cosmetic concerns.
Psychological: Anxiety about cancer risk, depression from chronic illness, body image issues, social isolation.
Social: Missed school/work for appointments and surgeries, financial burden of lifelong care, relationship challenges.
Management: Multidisciplinary approach including orthopedic surgery, oncology, physiotherapy, psychology, genetic counseling, and social work. Patient support groups valuable.
Postoperative Care
Post-Curettage Care
- Duration
- 0-2 weeks
- Key Activities
- Wound care, pain control, elevation
- Precautions
- Restrict weight-bearing if lower limb
- Duration
- 2-6 weeks
- Key Activities
- Gentle ROM exercises, edema control
- Precautions
- Avoid heavy lifting; protect surgical site
- Duration
- 6-12 weeks
- Key Activities
- Gradual return to activity, physiotherapy
- Precautions
- Serial X-rays to confirm graft incorporation
- Duration
- 12+ weeks
- Key Activities
- Return to full activities, sport
- Precautions
- Ongoing surveillance for recurrence
Hand Surgery Rehabilitation
- Week 1: Bulky dressing, elevation, finger ROM out of splint
- Week 2: Remove bulky dressing, begin active exercises
- Week 3-4: Gentle grip strengthening, scar massage
- Splinting: Volar resting splint at night only if needed
- Edema control: Coban wrap, elevation, retrograde massage
- Week 4-6: Progressive strengthening, putty exercises
- Week 6-8: Return to light work activities
- Week 8-12: Full return to sport/manual work
- Follow-up: Radiographs at 6 weeks, 3 months, 6 months
- Recurrence watch: Any pain, swelling, or new deformity
Weight-Bearing Progression
- Initial
- Sling comfort only
- Progression
- Immediate ROM
- Full WB
- 2-4 weeks
- Initial
- Touch-down WB
- Progression
- Progressive at 4-6 weeks
- Full WB
- 6-8 weeks
- Initial
- Non-weight bearing
- Progression
- TDWB at 6 weeks
- Full WB
- 8-12 weeks
- Initial
- Heel walking/cast boot
- Progression
- Progressive at 4 weeks
- Full WB
- 6-8 weeks
Surveillance Protocol
- Post-op X-ray: Immediate (baseline)
- 6 weeks: Confirm healing, graft incorporation
- 3 months: Assess for recurrence
- 6 months: Check remodeling
- Annually: Lifelong (malignancy surveillance)
- MRI: If pain or suspicious changes
- Wound healing: Complete by 2 weeks
- Pain: Should improve progressively
- Function: Full ROM by 6-8 weeks (hand)
- Red flags: Night pain, new mass, rapid growth
- Other lesions: Monitor all known enchondromas
Patients and families must understand that enchondromatosis requires LIFELONG follow-up even after successful surgery. New lesions can develop, existing lesions can recur, and malignant transformation remains a risk throughout life.
Outcomes
Overall Prognosis
Surgical Outcomes by Procedure
- Success Rate
- 95%+
- Recurrence
- 5-10%
- Key Outcomes
- Excellent function, minimal complications
- Success Rate
- 85-90%
- Recurrence
- 10-15%
- Key Outcomes
- Good results; may need repeat surgery
- Success Rate
- 80-90%
- Recurrence
- N/A
- Key Outcomes
- Deformity correction achieved; may lose correction with growth
- Success Rate
- 90%+
- Recurrence
- N/A
- Key Outcomes
- Predictable if timed correctly
- Success Rate
- 70-85%
- Recurrence
- N/A
- Key Outcomes
- Good outcomes but high complication rate (30-50%)
- Success Rate
- Variable
- Recurrence
- 10-20%
- Key Outcomes
- Depends on grade, margins, and metastatic status
Functional Outcomes
- Hand: Generally excellent after isolated curettage
- DASH scores: Near normal with single lesions
- Multiple lesions: May have residual weakness
- Work capacity: Usually maintained
- Fine motor: Preserved unless multiple digit involvement
- Ambulation: Most achieve community ambulation
- Leg length discrepancy: Managed with shoe raise or surgery
- Hip/Knee function: Depends on juxta-articular involvement
- Sport: Often modified activities recommended
- Walking aids: Required in 10-20% with severe disease
Quality of Life Factors
- Multiple surgeries (average 3-5 over lifetime)
- Chronic pain in 20-40% of patients
- Functional limitations in severe cases
- Cosmetic concerns (limb asymmetry, scars)
- Fatigue from chronic disease burden
- Cancer anxiety (lifelong malignancy risk)
- Body image issues (especially adolescents)
- Educational/vocational impact
- Healthcare burden (frequent appointments)
- Financial impact of lifelong care
Guidelines, Registries & Global Practice
Global Epidemiology
Enchondromatosis is rare and almost universally sporadic. Ollier disease has an estimated prevalence in the order of 1 in 100,000, and Maffucci syndrome is considerably rarer. Both are caused by post-zygotic somatic mosaic IDH1/IDH2 mutations and are therefore non-hereditary, with the same mutational spectrum seen worldwide (no consistent ethnic or geographic predilection). According to PubMed, in the largest international multicentre series (161 patients across 13 European centres) the cross-sectional incidence of secondary chondrosarcoma was 40%, rising with long-bone, flat-bone and especially pelvic involvement (Verdegaal et al, The Oncologist 2011) DOI. The defining IDH1/IDH2 mutation prevalence of 87% in enchondromas is consistent across populations (Pansuriya et al, Nature Genetics 2011) DOI.
Guideline & Society Guidance (Side-by-Side)
There is no disease-specific international guideline for enchondromatosis; management is extrapolated from cartilage-tumour and sarcoma frameworks. The table summarises how the major bodies frame the relevant cartilage-tumour pathway.
- Relevant Guidance
- Bone sarcoma guideline: suspected chondrosarcoma referred to a sarcoma reference centre before biopsy; grade 1 / atypical cartilaginous tumour of long bones may be managed by curettage, higher-grade by wide resection; chondrosarcoma is chemo/radio-resistant
- Evidence Level
- Expert consensus, graded recommendations
- Relevant Guidance
- Suspected sarcoma referral pathways and specialist MDT (sarcoma advisory group) review; imaging-led triage with biopsy only after staging at the treating centre
- Evidence Level
- Guideline-based pathway
- Relevant Guidance
- Bone cancer guideline: multidisciplinary management at sarcoma centres, wide excision for conventional chondrosarcoma, active surveillance or intralesional surgery for atypical cartilaginous tumour of the appendicular skeleton
- Evidence Level
- Category 2A consensus
- Relevant Guidance
- Centralised referral of suspected primary bone sarcoma to designated sarcoma services with MDT governance
- Evidence Level
- Service-standard consensus
Registry & High-Volume Evidence
- International EMSOS cohort (n=161): 40% chondrosarcoma incidence; pelvic disease OR 3.8 (Verdegaal 2011)
- Appendicular grade 1 series (n=113): 2.7% local recurrence after extended curettage + cryoadjuvant, MSTS 95% (El Masry 2022)
- Genetic cohorts: IDH1/IDH2 mutations in 87% of enchondromas (Pansuriya 2011)
- No dedicated national enchondromatosis registry exists; data come from sarcoma-centre cohorts and rare-disease networks
- Curettage vs wide resection for grade 1 lesions varies by centre and remains debated, but appendicular tumours are increasingly managed by joint-sparing surgery
- Surveillance intensity is non-standardised; most centres favour risk-stratified imaging (more aggressive for pelvic/long-bone and Maffucci disease)
- Genetic testing is performed on affected tissue (not blood), reflecting somatic mosaicism; genetic counselling reinforces the sporadic, non-hereditary nature of the disease
- Centralisation: high-income systems centralise sarcoma care, with diagnosis typically established at paediatric centres and lifelong surveillance coordinated through tertiary sarcoma services and multidisciplinary tumour boards; access and timeliness vary in lower-resourced settings
- Patient support: rare-disease and limb-difference support organisations provide patient resources alongside psychological support
MCQ Practice Points
Q: What is the difference between Ollier disease and Maffucci syndrome?
A: Both are non-hereditary enchondromatosis syndromes. Ollier disease: Multiple enchondromas with asymmetric distribution, typically unilateral predominance; No associated soft tissue lesions. Maffucci syndrome: Multiple enchondromas PLUS soft tissue hemangiomas (venous malformations, phleboliths on X-ray). Maffucci has higher malignancy risk (40-50% vs 25-30% for Ollier). Both present in childhood with limb deformity, shortening, and pathological fractures.
Q: What is the malignancy risk in Ollier disease and how do you monitor for malignant transformation?
A: Lifetime chondrosarcoma risk is 25-30% (much higher than solitary enchondroma which is less than 1%). Warning signs for transformation: New or increasing pain (especially at rest); Rapid growth on serial imaging; Size greater than 5cm; Soft tissue mass on MRI; Cortical destruction. Surveillance: Clinical review annually; Imaging of symptomatic lesions; Low threshold for biopsy/resection of suspicious lesions. Transformation usually occurs in adulthood (3rd-4th decade).
Q: What are the clinical features and natural history of Ollier disease?
A: Presents in early childhood (first decade) with limb shortening, angular deformity, and palpable bony swelling. Typically asymmetric distribution, often with unilateral predominance. Common sites: hands, feet, long bones. Complications: Pathological fractures (heal normally); Progressive deformity; Limb length discrepancy. Natural history: Lesions may stabilize after skeletal maturity but remain at risk for malignant transformation throughout life.
Q: What is the typical imaging appearance of enchondromas in Ollier disease?
A: Multiple well-defined lytic lesions with chondroid matrix (rings and arcs calcification). Distribution: Metaphyseal, extending toward physis in immature skeleton. May cause expansion and cortical thinning without destruction. Characteristic: Streaky or columnar appearance extending from physis (reflecting origin from growth plate cartilage). MRI: High T2 signal (cartilage), lobular architecture. CT best for matrix calcification and cortical integrity assessment.
Q: How is limb deformity and length discrepancy managed in Ollier disease?
A: Conservative: Shoe lifts for mild LLD (less than 2cm). Surgical options: (1) Epiphysiodesis of contralateral limb for moderate LLD; (2) Lengthening procedures (distraction osteogenesis) for severe LLD; (3) Corrective osteotomy for angular deformity; (4) Curettage and grafting for symptomatic lesions. Timing: Defer elective surgery until skeletal maturity if possible due to high recurrence risk in immature skeleton. Amputation rarely needed but considered for severe, recurrent deformity.
At a Glance
Enchondromatosis encompasses multiple enchondroma syndromes with high malignant transformation risk. Ollier disease features multiple unilateral enchondromas with 25-30% chondrosarcoma transformation by age 40. Maffucci syndrome combines enchondromas with soft tissue hemangiomas and carries nearly 100% lifetime malignancy risk—both chondrosarcoma (from cartilage) and angiosarcoma (from hemangiomas). Both are caused by somatic IDH1/IDH2 mutations (87%)—sporadic, not inherited. Lifelong surveillance is mandatory: annual clinical examination, low threshold for MRI of any symptomatic lesion, and urgent biopsy consideration for pain, growth, or soft tissue mass. Common skeletal complications include limb length discrepancy, angular deformity, and pathological fractures.
OLLIEROllier Disease Features
Hook:OLLIER disease = One-sided Lesions with high malignancy Risk!
MAFFUCCIMaffucci Syndrome Red Flags
Hook:MAFFUCCI syndrome = Multiple tumors with Frightening malignancy risk!
WATCHSurveillance Protocol - WATCH
Hook:WATCH closely for malignant transformation - patient's life depends on it!
Exam Viva Scenarios
Practise clinical reasoning and management decisions out loud
“A 7-year-old girl presents with limb length discrepancy (left leg 3cm shorter than right). X-rays show multiple enchondromas in the left femur, tibia, and foot. Right leg is normal. Parents ask about diagnosis and prognosis. What is your diagnosis and how do you counsel the family?”
“A 32-year-old woman with known Ollier disease presents with 3 months of progressive right groin pain. She has multiple enchondromas in the right femur documented since childhood. X-ray shows 8cm lesion in proximal femur with cortical thinning. MRI shows heterogeneous T2 signal with small soft tissue component. How do you proceed?”
“A 28-year-old man with Maffucci syndrome (multiple enchondromas plus hemangiomas) presents for annual surveillance. He is asymptomatic. Physical exam reveals stable enchondromas in hands and feet, but one hemangioma on his left calf has doubled in size over 6 months and feels firm. How do you manage this patient?”
Key Definitions
- **Ollier disease**: Multiple unilateral enchondromas, sporadic, 25-30% malignancy risk
- **Maffucci syndrome**: Enchondromas PLUS hemangiomas, nearly 100% malignancy risk
- **IDH1/IDH2 somatic mutations**: Found in 87% of lesions, NOT inherited
- **Chondrosarcoma**: Malignant cartilage tumor from enchondroma transformation
Clinical Presentation
- Childhood: limb deformity, limb length discrepancy, palpable masses
- Adolescence: pathological fractures, progressive deformities
- Adulthood: pain in lesion (RED FLAG for malignancy), soft tissue mass
- Maffucci: visible hemangiomas (soft compressible masses)
Diagnosis
- **Skeletal survey**: Document all lesions at baseline (X-rays all limbs)
- **MRI**: For large proximal lesions and any symptomatic lesion
- **X-ray features**: Multiple enchondromas, rings-and-arcs calcification, unilateral
- **Genetic testing**: IDH1/IDH2 mutation testing available but not routine
Red Flags for Malignancy
- **Pain without trauma** - most important clinical sign (90% sensitivity)
- Progressive enlargement on serial imaging
- Soft tissue mass on MRI (highly specific for chondrosarcoma)
- Cortical breakthrough on CT, lesion size over 5cm
Surveillance Protocol
- **Annual clinical exam**: Check for pain, masses, deformities
- **Skeletal survey**: Repeat every 2-3 years in childhood
- **Low threshold for MRI**: Any painful lesion gets MRI immediately
- **Biopsy if suspicious**: CT-guided with excisable trajectory
Management of Deformities
- **Limb length discrepancy**: Epiphysiodesis (2-5cm) or lengthening (over 5cm)
- **Angular deformity**: Guided growth or corrective osteotomy
- **Pathological fracture**: Immobilize, heal, delayed curettage and graft
- **MRI before elective surgery**: Rule out malignancy before deformity correction
Malignancy Treatment
- **Chondrosarcoma**: Wide excision with 5-10mm margins (curettage inadequate)
- **Reconstruction**: Allograft, endoprosthesis, or allograft-prosthetic composite
- **Angiosarcoma (Maffucci)**: Wide excision, often requires amputation
- **No chemo/radiation**: Chondrosarcoma does NOT respond; surgery only curative
Prognosis
- **Ollier malignancy risk**: 25-30% develop chondrosarcoma by age 40
- **Maffucci malignancy risk**: Nearly 100% lifetime (chondrosarcoma + angiosarcoma)
- **Chondrosarcoma survival**: 90% 5-year for Grade 1 with wide excision
- **Surveillance benefit**: Early detection enables limb salvage and better survival
Evidence Base and Key Studies
Discovery of IDH Mutations in Enchondromatosis
- Somatic heterozygous IDH1 (R132C, R132H) or IDH2 (R172S) mutations identified in 87% of enchondromas and 70% of spindle cell haemangiomas
- Across the cohort, 81% of Ollier disease and 77% of Maffucci syndrome subjects carried IDH1 (98%) or IDH2 (2%) mutations in their tumours
- Immunohistochemistry suggested intraneoplastic and somatic mosaicism, explaining the sporadic, non-hereditary, mosaic distribution
- IDH1 mutations were associated with DNA hypermethylation and downregulated gene expression (epigenetic dysregulation)
- Mutations were also detected in 40% of solitary central cartilage tumours and in chondrosarcoma cell lines
Incidence and Predictors of Chondrosarcoma in Ollier/Maffucci
- International multicentre study of 161 patients (144 Ollier, 17 Maffucci) from 13 European centres and one national databank
- Overall observed incidence of secondary chondrosarcoma was 40% (likely higher as a lifelong, age-dependent risk)
- Risk varied by distribution: 15% for hand/foot-only disease (group I) versus 43-46% when long/flat bones were involved (groups II-III)
- Pelvic enchondromas markedly increased chondrosarcoma risk (odds ratio 3.8, p = 0.001)
- Patients with long-bone or axial (especially pelvic) enchondromas were identified as the group needing regular screening
Maffucci Syndrome - Functional and Neoplastic Significance
- Case report and review of the world literature on Maffucci syndrome from its original description onward
- Documented frequent malignant transformation, including chondrosarcoma arising from enchondromas
- Highlighted association with non-skeletal malignancies including ovarian neoplasms and other primary tumours
- Emphasised the markedly elevated lifelong malignancy risk that distinguishes Maffucci from Ollier disease
- Reinforced that spindle cell (cutaneous/soft tissue) haemangiomas are the pathognomonic feature of Maffucci syndrome
Curettage and Adjuvant for Low-Grade (Grade 1) Chondrosarcoma
- Retrospective series of 113 patients with low-grade (grade 1 / atypical cartilaginous) chondrosarcoma of the appendicular skeleton
- Treated with extended curettage, liquid-nitrogen cryoadjuvant, polymethylmethacrylate cement filling and prophylactic fixation
- Local recurrence occurred in only 3 patients (2.7%) at a mean follow-up of 110 months
- Non-oncological complications in 5.3%; mean MSTS functional score 95% with no metastases or disease-related mortality
- Grade 1 chondrosarcoma (in non-syndromic appendicular sites) can be controlled with joint-sparing intralesional surgery
Metachondromatosis is Caused by Germline PTPN11 Mutations
- Whole-genome sequencing of a single proband plus linkage analysis identified the metachondromatosis gene
- An 11-bp frameshift deletion in exon 4 of PTPN11 segregated with the phenotype
- A second family carried an independent exon 4 nonsense mutation, confirming PTPN11 loss-of-function as causal
- No protein-truncating PTPN11 variants were found in 469 controls, supporting pathogenicity
- Metachondromatosis (OMIM 156250) is an autosomal dominant disorder, genetically distinct from IDH-driven Ollier/Maffucci
Ivosidenib (Mutant IDH1 Inhibitor) in Advanced Chondrosarcoma
- Phase I multicentre dose-escalation/expansion study of oral ivosidenib monotherapy in 21 patients with advanced IDH1-mutant chondrosarcoma
- Toxicity was mostly grade 1-2; only one grade 3 or higher event (hypophosphataemia) was treatment related
- Plasma 2-hydroxyglutarate fell substantially in all patients (14-94%), to levels seen in healthy individuals
- Median progression-free survival was 5.6 months with a 6-month PFS rate of 39.5%
- 11 of 21 patients (52%) achieved stable disease, supporting targeting of the IDH/2-HG axis