Aggressive Sarcoma | 50% NF1-Associated | Worst Sarcoma Prognosis
- MPNST has worst prognosis of all soft tissue sarcomas - 5-year survival 40-60%
- 50% occur in NF1 patients from plexiform neurofibroma transformation (25-30% lifetime risk)
- PET-CT SUV greater than 3.5 has 89% sensitivity and 95% specificity for MPNST in NF1
- Wide surgical excision with 2 cm margins en bloc with nerve is only curative treatment
- Adjuvant radiotherapy improves local control 60-70% to 80-85% but NOT overall survival
- “NF1-associated MPNST has WORSE prognosis than sporadic (21% vs 42% 5-year survival)
- “S100 positive only 50-70% and FOCAL (not diffuse like schwannoma) - negative does not exclude
- “PRC2 complex mutations SUZ12/EED in 70-90% - H3K27me3 loss is diagnostic marker
- “Rapid growth, severe pain 70%, neurological deficit - distinguish from stable neurofibroma
- “Chemotherapy has modest benefit - response rate 20-30% lower than other sarcomas
50% occur in NF1 patients, 21% 5-year survival - Plexiform neurofibroma transformation in 25-30% lifetime risk. NF1-MPNST has significantly worse prognosis than sporadic MPNST (21% vs 42%). Median age 26 years in NF1 vs 40-50 sporadic.
SUV greater than 3.5: 89% sensitivity, 95% specificity - Critical tool for detecting malignant transformation in NF1. SUV less than 2.5 has 100% negative predictive value. Intermediate 2.5-3.5 needs close surveillance or biopsy.
Wide excision 2 cm margins en bloc with nerve - Only curative treatment. R0 resection: 65% 5-year survival vs 30% for incomplete resection. Nerve sacrifice mandatory. Amputation if neurovascular bundle encased.
Radiotherapy improves local control NOT survival - 60-66 Gy adjuvant radiation improves local control from 60-70% to 80-85% but no overall survival benefit. Chemotherapy response rate 20-30% (lower than other sarcomas).
MPNSTMPNST Key Features
Hook:MPNST - remember the key molecular and clinical features that define this aggressive tumor!
HIGH RISKHIGH RISK Features Suggesting MPNST
Hook:HIGH RISK - features that should trigger immediate biopsy and PET-CT in NF1 patients!
PETPET-CT SUV Interpretation
Hook:PET thresholds - under 2.5 reassuring, over 3.5 urgent action, 2.5-3.5 watch closely!
Overview and Epidemiology
Malignant peripheral nerve sheath tumor (MPNST) is an aggressive soft tissue sarcoma arising from cells of the peripheral nerve sheath. It represents 5-10% of all soft tissue sarcomas but carries one of the worst prognoses among sarcomas. Approximately 50% of cases occur in patients with neurofibromatosis type 1 (NF1), typically arising from malignant transformation of plexiform neurofibromas.
Epidemiology
- Annual incidence general population: 0.001% (1 per 100,000)
- Annual incidence in NF1 patients: 0.16% overall, 2-5% in adults with plexiform neurofibroma
- Lifetime risk in NF1: 8-13% overall population, 25-30% in those with plexiform neurofibroma
- Age: Median 26-30 years in NF1 patients, 40-50 years in sporadic
- Gender: Slight male predominance (1.2:1)
- Proximal extremities: 40-50% (thigh most common, upper arm)
- Trunk: 25-30% (paraspinal, retroperitoneum)
- Head and neck: 15-20%
- Distal extremities: 5-10%
- Brachial plexus and lumbosacral plexus commonly involved in NF1
Etiological Classification
- Frequency
- 50%
- Median Age
- 26-30 years
- Pathogenesis
- Plexiform neurofibroma transformation, germline NF1 plus somatic TP53
- 5-Year Survival
- 21%
- Frequency
- 40%
- Median Age
- 40-50 years
- Pathogenesis
- Somatic NF1 inactivation, TP53, PRC2 mutations
- 5-Year Survival
- 42%
- Frequency
- 10%
- Median Age
- Variable
- Pathogenesis
- Prior radiation over 40 Gy, latency 10-20 years
- 5-Year Survival
- 20-30%
NF1-associated MPNST has significantly worse prognosis than sporadic disease.
Pathophysiology and Molecular Pathogenesis
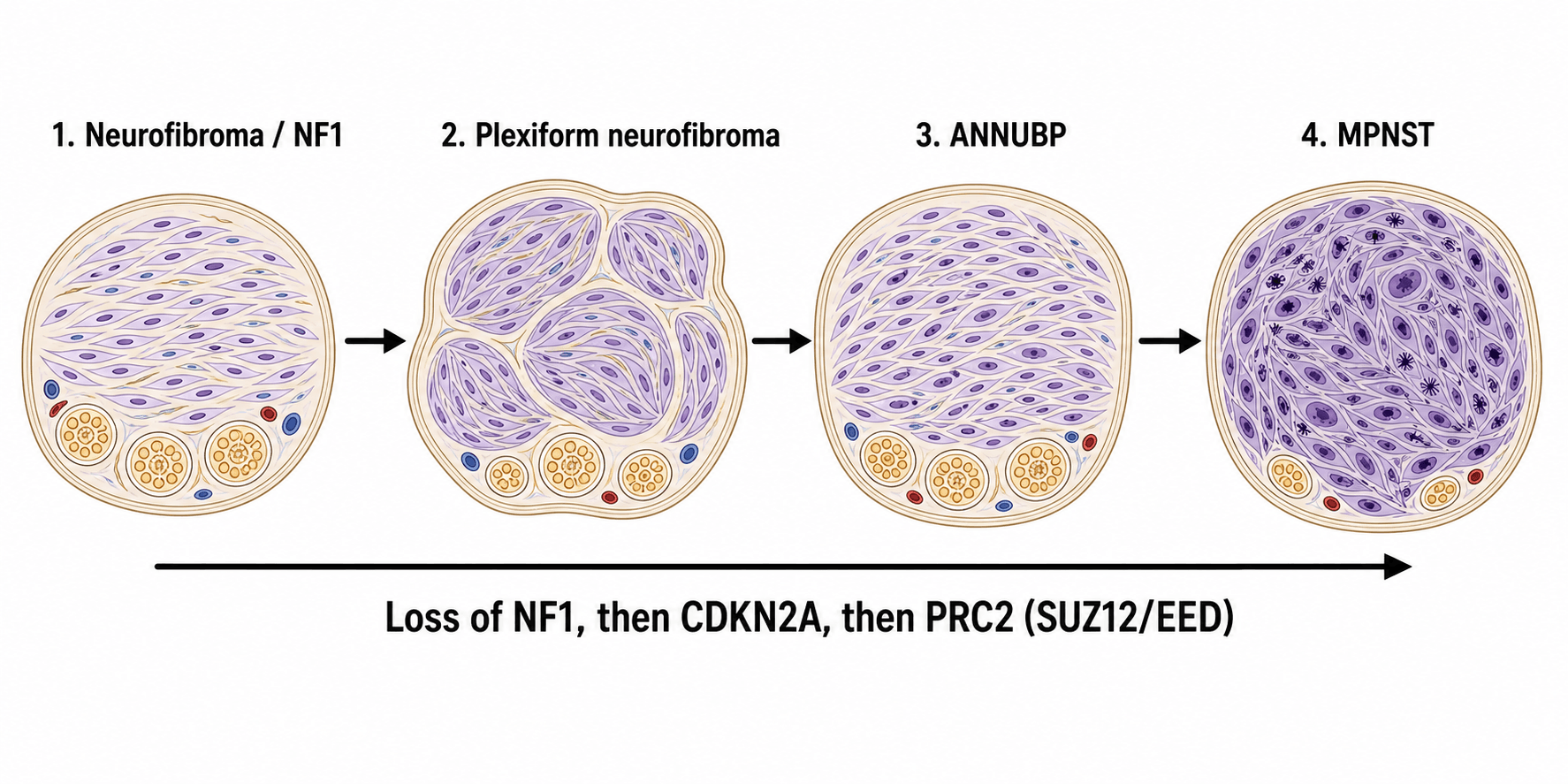
Genetic Pathways
NF1-Associated MPNST (Multistep Progression):
The pathway from neurofibroma to MPNST involves sequential genetic hits:
Malignant Transformation Pathway in NF1
Germline NF1 mutation plus somatic second hit (loss of heterozygosity).
Characteristics: Slow-growing, soft, painless, stable for years. Low cellularity, wavy spindle cells.
Additional genetic changes: Increased cellularity, mild atypia, hypercellularity.
Still benign but at higher risk for progression. Difficult to diagnose histologically.
TP53 inactivation (75% of MPNST) plus PRC2 complex mutations (SUZ12 or EED loss in 70-90%).
Also CDKN2A deletion in 50-60%. Result: Aggressive malignant transformation.
- NF1 loss: Neurofibromin deficiency causes Ras-MAPK hyperactivation, uncontrolled proliferation
- TP53 loss: Eliminates cell cycle checkpoints and apoptosis, allows accumulation of mutations
- PRC2 loss (SUZ12/EED): Epigenetic dysregulation, loss of H3K27 trimethylation marker
- CDKN2A deletion: Loss of p16 tumor suppressor, cell cycle dysregulation
- Somatic biallelic NF1 inactivation (not inherited)
- TP53 mutations common
- PRC2 mutations (SUZ12, EED)
- CDKN2A deletions
- No prior neurofibroma or NF1 diagnosis
- Median latency 10-20 years after radiation exposure
- Radiation doses typically greater than 40 Gy
- Field includes peripheral nerve
- Often higher grade, worse prognosis
- Examples: Post-treatment for breast cancer, lymphoma, childhood malignancies
Risk Factors for Malignant Transformation in NF1
- Risk Level
- High
- Transformation Rate
- 25-30% lifetime
- Management
- Annual surveillance, PET-CT if concerning
- Risk Level
- High
- Transformation Rate
- Higher than superficial
- Management
- MRI surveillance, low threshold for PET-CT
- Risk Level
- Moderate
- Transformation Rate
- Difficult to monitor
- Management
- Regular imaging, PET-CT surveillance
- Risk Level
- Low
- Transformation Rate
- Rare transformation
- Management
- Clinical surveillance only
Internal plexiform neurofibromas in NF1 patients have the highest malignant potential.
Histopathology and Classification
Macroscopic Features
Gross Appearance:
- Size: Usually greater than 5 cm at diagnosis (mean 8-10 cm)
- Margins: Poorly defined, infiltrative (unlike encapsulated schwannoma)
- Cut surface: Tan-gray, fleshy, heterogeneous
- Necrosis: Common in high-grade tumors (50-70%)
- Hemorrhage: Frequent
- Nerve origin: May be visible in smaller tumors, obscured in large masses
Microscopic Features
High-Grade MPNST (90% of cases)
- Dense fascicles of spindle cells in whorled pattern
- "Marbled" alternating areas of dense cellularity and myxoid zones
- Perivascular accentuation (cells cluster around vessels)
- Infiltrative margins into surrounding tissue
- Spindle cells with hyperchromatic nuclei
- Moderate to severe nuclear pleomorphism
- High nuclear-to-cytoplasmic ratio
- Wavy, buckled nuclei (similar to benign Schwann cells)
- High mitotic index: typically greater than 10 mitoses per 10 HPF
- Atypical mitotic figures common
- Geographic necrosis in 50-70%
- Predictor of aggressive behavior
Most MPNST are high-grade at diagnosis.
Histological Variants
- 5-10% of MPNST
- Contains malignant skeletal muscle component
- Desmin, myogenin, MyoD1 positive in rhabdomyoblastic areas
- Worse prognosis than conventional MPNST
- Epithelioid cytology rather than spindle
- More common in superficial locations
- S100 often positive
- Must distinguish from melanoma, epithelioid sarcoma
Differential Diagnosis
MPNST is a spindle-cell sarcoma that overlaps morphologically with several other neoplasms. Immunohistochemistry and clinical context are key to distinguishing them.
- Key Distinguishing Feature
- Low mitotic rate, no necrosis, retains uniform architecture
- Immunohistochemistry
- S100 diffuse, H3K27me3 RETAINED
- Behaviour
- Benign / pre-malignant (atypical)
- Key Distinguishing Feature
- Encapsulated, Antoni A/B, Verocay bodies, hyalinised vessels
- Immunohistochemistry
- S100 STRONG and DIFFUSE, SOX10 diffuse
- Behaviour
- Benign
- Key Distinguishing Feature
- Uniform spindle cells, SS18-SSX fusion
- Immunohistochemistry
- Cytokeratin / EMA focal positive, TLE1 positive, S100 focal
- Behaviour
- Malignant
- Key Distinguishing Feature
- Herringbone pattern, no nerve origin
- Immunohistochemistry
- S100 negative, CD34 (DFSP), H3K27me3 retained
- Behaviour
- Malignant (variable grade)
- Key Distinguishing Feature
- Blunt-ended (cigar) nuclei, fascicles at right angles
- Immunohistochemistry
- SMA / desmin / h-caldesmon positive, S100 negative
- Behaviour
- Malignant
- Key Distinguishing Feature
- Junctional component, history of pigmented lesion
- Immunohistochemistry
- S100 diffuse, SOX10 diffuse, H3K27me3 RETAINED
- Behaviour
- Malignant
Key discriminators: diffuse strong S100 favours schwannoma or melanoma over MPNST (which shows focal/patchy S100 in only 50-70%); H3K27me3 loss favours MPNST and is retained in melanoma, neurofibroma and most mimics; epithelial markers (cytokeratin, EMA, TLE1) point to synovial sarcoma; and smooth-muscle markers point to leiomyosarcoma.
Grading System
- Score 1
- Well-differentiated
- Score 2
- Moderately differentiated
- Score 3
- Poorly differentiated
- Score 1
- 0-9 per 10 HPF
- Score 2
- 10-19 per 10 HPF
- Score 3
- 20 or more per 10 HPF
- Score 1
- None
- Score 2
- Less than 50%
- Score 3
- 50% or greater
Total Score:
- Grade 1 (low): 2-3 points (rare in MPNST, under 10%)
- Grade 2 (intermediate): 4-5 points (20%)
- Grade 3 (high): 6-8 points (70%)
Most MPNST are high-grade (Grade 3) at diagnosis.
Atypical Neurofibromatous Neoplasm of Uncertain Biologic Potential (ANNUBP)
The transformation timeline above relies on an intermediate lesion labelled "atypical neurofibroma," the differential table lists "cellular / atypical neurofibroma," and the low-grade MPNST discussion notes the difficulty of separating the two — but this premalignant precursor deserves to be named and defined, because it is the diagnostic grey zone that determines whether a lesion is watched, marginally excised, or treated as a sarcoma. (The benign neurofibroma itself is covered in the dedicated neurofibroma topic; here the focus is the precursor on the road to MPNST.)
The consensus term. A 2017 international consensus replaced the vague "atypical neurofibroma" with atypical neurofibromatous neoplasm of uncertain biologic potential (ANNUBP) — the histological and biological bridge between a benign neurofibroma and MPNST. It is a recognised premalignant lesion, especially within large internal/plexiform neurofibromas in NF1.
Diagnostic criteria. ANNUBP is diagnosed when at least two of four features are present:
- Cytologic atypia
- Loss of the normal neurofibroma architecture
- Hypercellularity
- Low-level mitotic activity — present but below the MPNST threshold (fewer than 3 mitoses per 10 high-power fields), with no necrosis
The distinction from low-grade MPNST is essentially quantitative: a higher mitotic count (3 or more per 10 HPF) and/or necrosis push a lesion into MPNST. This is a genuinely difficult call that requires expert sarcoma pathology.
Molecular correlate. Progression from neurofibroma to ANNUBP is marked by biallelic CDKN2A (p16) inactivation — loss of p16 by immunohistochemistry supports the diagnosis and dovetails with the CDKN2A deletions seen in established MPNST. Importantly, H3K27me3 is usually retained at the ANNUBP stage; its global loss is a later event, occurring with PRC2 inactivation as the lesion becomes MPNST. So an atypical lesion that has lost H3K27me3 should raise concern for transformation.
Clinical relevance and management. ANNUBP often corresponds to a focally concerning area within a plexiform neurofibroma — for example a region of intermediate PET avidity (SUVmax 2.5–3.5) or new growth — and may be what a targeted biopsy of that area yields. Unlike MPNST, ANNUBP is managed by complete (marginal) excision with clear margins alone: it does not require the wide 2 cm margins, nerve sacrifice, radiotherapy or chemotherapy used for MPNST, but it does mandate negative margins and ongoing surveillance because of its malignant potential.
The intermediate lesion in NF1 transformation is ANNUBP (atypical neurofibromatous neoplasm of uncertain biologic potential): at least 2 of cytologic atypia / architectural loss / hypercellularity / low-level mitoses (fewer than 3 per 10 HPF), no necrosis, CDKN2A/p16 loss but retained H3K27me3. Manage by complete excision with clear margins — wide margins, radiotherapy and chemotherapy belong to MPNST, not ANNUBP. Recognising ANNUBP explains the intermediate-SUV lesion and the "can't tell from low-grade MPNST" trap.
Investigations and Staging
Imaging
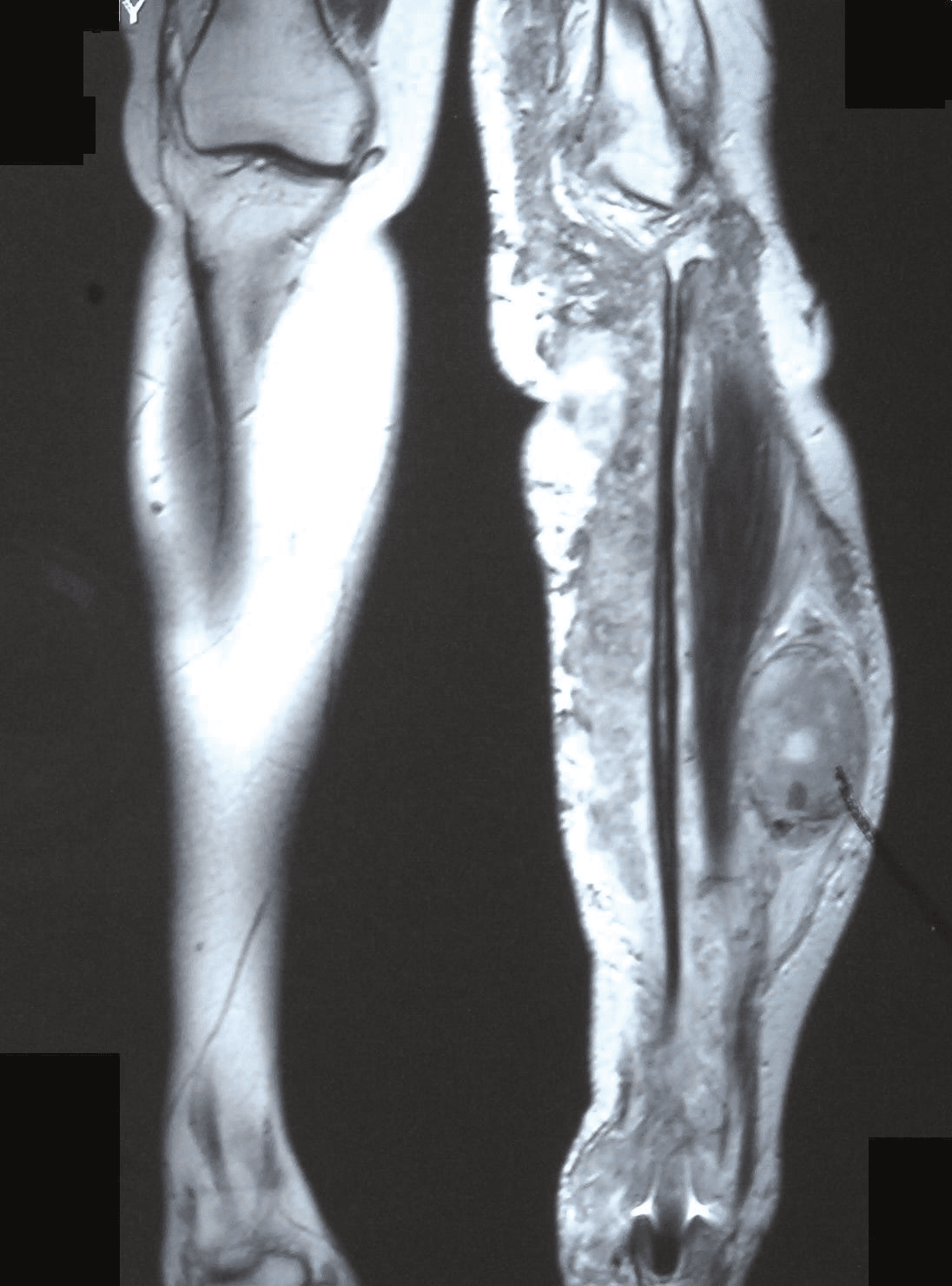
MRI - Gold Standard for Local Staging
- T1-weighted: Anatomical detail
- T2-weighted with fat suppression: Tumor extent, edema
- T1 post-gadolinium with fat suppression: Enhancement pattern
- Include entire compartment plus joint above and below
- Isointense to muscle
- Heterogeneous signal (necrosis, hemorrhage)
- Irregular, infiltrative margins (unlike well-defined benign tumors)
- Loss of fat planes surrounding tumor
- Heterogeneous signal (areas of hyperintensity and hypointensity)
- Loss of uniform T2 hyperintensity seen in benign neurofibromas
- Central necrosis: Fluid signal
- Peritumoral edema in aggressive tumors
- Heterogeneous enhancement
- Non-enhancing necrotic areas
- Rim enhancement around necrosis
- Enhancement of involved nerve proximally and distally
- Benign Neurofibroma
- Usually under 5 cm
- MPNST
- Greater than 5 cm (mean 8-10 cm)
- Benign Neurofibroma
- Uniform T2 hyperintense
- MPNST
- Heterogeneous signal
- Benign Neurofibroma
- Well-defined
- MPNST
- Irregular, infiltrative
- Benign Neurofibroma
- Absent
- MPNST
- Present 50-70%
- Benign Neurofibroma
- Present (central low, peripheral high T2)
- MPNST
- Lost
- Benign Neurofibroma
- Minimal
- MPNST
- Perilesional edema common
MRI guides surgical resection planning and biopsy location.
Biopsy
Critical oncological principles:
- Image-guided core needle biopsy PREFERRED (14-16 gauge, multiple cores 4-6)
- Biopsy tract MUST be in line with planned surgical incision (will be excised en bloc)
- Target solid areas, avoid necrosis (image guidance essential)
- NEVER perform excisional biopsy for suspected MPNST (violates oncologic principles)
- Send for H&E, immunohistochemistry (S100, SOX10, desmin, cytokeratin, Ki67, H3K27me3), and FISH/molecular if uncertain
Referral to sarcoma center before biopsy is ideal for optimal outcomes.
Pathology Assessment Required:
- FNCLCC grade (critical for prognosis and adjuvant therapy decisions)
- S100 status (remember: Only 50-70% positive)
- Ki67 proliferation index
- H3K27me3 status (loss suggests PRC2 mutation, supports MPNST diagnosis)
- Rule out other spindle cell sarcomas (synovial, fibrosarcoma)
AJCC Staging
- T (Size/Depth)
- T1 (5 cm or less) superficial or deep
- Grade
- Grade 1 (low)
- 5-Year Survival
- 90%
- T (Size/Depth)
- T2-T4 (greater than 5 cm)
- Grade
- Grade 1 (low)
- 5-Year Survival
- 80%
- T (Size/Depth)
- T1 (5 cm or less)
- Grade
- Grade 2-3 (high)
- 5-Year Survival
- 70%
- T (Size/Depth)
- T2 (greater than 5 cm but 10 cm or less)
- Grade
- Grade 2-3 (high)
- 5-Year Survival
- 60%
- T (Size/Depth)
- T3-T4 (greater than 10 cm)
- Grade
- Grade 2-3 (high)
- 5-Year Survival
- 40-50%
- T (Size/Depth)
- Any T with N1 or M1
- Grade
- Any grade
- 5-Year Survival
- 10-15%
Most MPNST present as Stage IIIB (large, deep, high-grade) or Stage IV (metastatic).
Clinical Assessment
History
Typical Presentation:
- Rapidly enlarging mass over weeks to months (KEY distinguishing feature from benign)
- Severe pain: 60-70% (neurogenic pain in nerve distribution)
- Prior history of stable neurofibroma with sudden growth (NF1 patients)
- Progressive neurological deficit: 30-40% (motor weakness, sensory loss)
- Duration: Usually less than 1 year of symptoms
NF1 Patients - Red Flags for Transformation:
- Rapid enlargement over weeks to months
- Change from soft to firm consistency
- New onset severe pain (previously painless)
- Neurological deficit (weakness, numbness)
- Weight loss, fatigue (10-20%)
- Suggests metastatic disease if present
- Rare in localized disease
- Warrant full staging workup
Physical Examination
- Large, firm mass (typically greater than 5 cm)
- Fixed to underlying structures (infiltrative)
- Overlying skin: Normal or tethered/ulcerated (advanced)
- Venous engorgement over mass (large tumors)
- Firm to hard consistency (versus soft neurofibroma - critical distinguishing feature)
- Non-mobile, fixed to deep tissues
- Poorly defined margins
- Tender in 60-70%
- Pulsation absent (distinguishes from vascular lesion)
- Motor deficit in distribution of involved nerve: 30-40%
- Sensory deficit: 40-50%
- Muscle atrophy if chronic
- Reduced or absent deep tendon reflexes
- Document pre-operative neurological status for informed consent
- Lymphadenopathy: Rare (MPNST rarely metastasizes to lymph nodes, less than 5%)
- Brachial or lumbosacral plexus involvement: Weakness and sensory loss in multiple nerve distributions
- Size greater than 5 cm
- Deep location (subfascial)
- Rapid growth (weeks to months)
- Fixed to underlying structures
- Neurological deficit
Management Algorithm
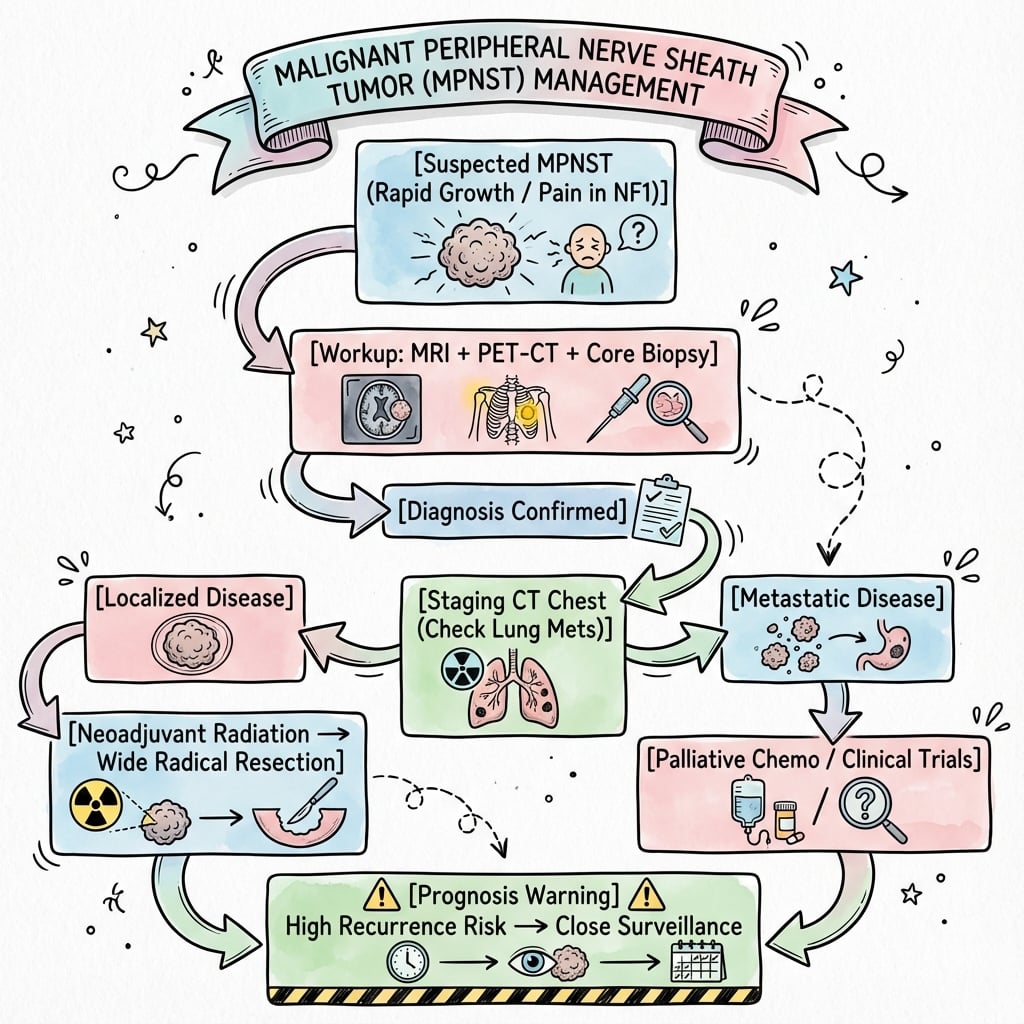
- 1
- 2
- 3
- 4
- 5
- 6
- 7
Multidisciplinary sarcoma team management is mandatory for optimal outcomes.
Adjuvant and Systemic Therapy
Radiotherapy
- High-grade MPNST (most cases, 90%)
- Positive or close margins (less than 1 mm)
- Large tumors (greater than 5 cm)
- Deep location (subfascial)
- Dose: 50 Gy in 25 fractions
- Advantages: Smaller treatment volume, potentially better local control
- Disadvantages: Wound complications (30-40%), delay to surgery (6-8 weeks post-RT)
- Dose: 60-66 Gy in 30-33 fractions
- Advantages: Immediate surgery, lower wound complications (15-20%)
- Disadvantages: Larger treatment volume (includes entire surgical bed)
- Additional 10-16 Gy to positive margin areas if re-resection not possible
- Improves local control from 60-70% to 80-85%
- Does NOT improve overall survival
- Recommended for most MPNST cases given high local recurrence risk
Chemotherapy
- Indication
- Initially unresectable, downstaging
- Regimen
- Ifosfamide plus doxorubicin
- Response Rate
- 20-30%
- Survival Benefit
- No proven benefit
- Indication
- Controversial, young patients high-grade
- Regimen
- Ifosfamide plus doxorubicin
- Response Rate
- N/A
- Survival Benefit
- No proven benefit
- Indication
- Palliative for symptomatic metastases
- Regimen
- Ifosfamide plus doxorubicin OR gemcitabine plus docetaxel
- Response Rate
- 20-40%
- Survival Benefit
- Median 8-12 months
Key Point: Chemotherapy has modest benefit in MPNST with response rates (20-30%) lower than other sarcomas (40-50%). NF1-associated MPNST particularly resistant to chemotherapy.
Novel Therapies
- MEK inhibitors (selumetinib, trametinib): Target Ras-MAPK pathway, modest activity (response under 10%)
- EZH2 inhibitors (tazemetostat): Target PRC2 pathway, limited single-agent activity
- Combination strategies under investigation
- Checkpoint inhibitors (anti-PD1, anti-CTLA4): Limited data, low response rates (under 10%)
- Tumor mutational burden typically low
No targeted therapy or immunotherapy is standard of care. Clinical trial enrollment encouraged.
Targeting the Precursor: MEK Inhibition in Plexiform Neurofibroma (Not MPNST)
MEK inhibitors are listed above as having minimal activity against established MPNST — but their real, practice-changing role is in the benign precursor, and the distinction is a high-yield one. Selumetinib, an oral MEK1/2 inhibitor that blocks the Ras–MAPK pathway driven by NF1 loss, is approved for paediatric NF1 patients with symptomatic, inoperable plexiform neurofibromas. In the pivotal SPRINT phase II trial, 35 of 50 children (70%) had a confirmed partial response (a reduction of at least 20% in tumour volume), most responses were durable, and there were meaningful improvements in pain, function and quality of life (Gross et al., N Engl J Med 2020, PMID 32187457).
The clinical logic — and the exam point — is the two-compartment distinction:
- In the benign plexiform neurofibroma, MEK inhibition can shrink the tumour, relieve symptoms and reduce the at-risk substrate.
- In established MPNST, the same drugs (and other targeted/immune agents) have minimal activity (response under 10%); complete surgical resection remains the only curative modality.
The danger this creates is mistaking a transforming or frankly malignant mass for a benign plexiform neurofibroma and "managing it medically." A plexiform neurofibroma that grows, becomes painful, develops a focally intermediate-to-high SUV, or shows worrying MRI change must be biopsied to exclude ANNUBP/MPNST before it is attributed to benign disease and offered a MEK inhibitor. (Detailed medical management of plexiform neurofibroma in NF1 belongs to the neurofibromatosis topic; the point here is the boundary with sarcoma.)
Selumetinib (a MEK inhibitor) is approved for symptomatic, inoperable plexiform neurofibroma in NF1 (SPRINT trial: ~70% partial responses), where it shrinks the benign precursor. It is not a treatment for established MPNST, where systemic agents respond poorly and surgery is the only cure. Before attributing an enlarging NF1 mass to benign disease and starting medical therapy, exclude transformation (ANNUBP/MPNST) with PET-directed biopsy.
Surgical Technique
Preoperative Assessment
- Sarcoma surgeon (orthopaedic oncology or surgical oncology)
- Medical oncologist
- Radiation oncologist
- Musculoskeletal radiologist
- Pathologist with sarcoma expertise
- NF1 specialist (if applicable)
- Review MRI to assess:
- Tumor extent and neurovascular involvement
- Relationship to major structures (vessels, bone, joints)
- Feasibility of limb salvage versus amputation
- Assess functional impact of nerve resection
- Discuss reconstruction options (nerve, soft tissue, bone if needed)
- Vascular surgery backup if major vessel involvement anticipated
- Anesthesia evaluation
- Nature of MPNST and poor prognosis (5-year survival 40-60%)
- Nerve sacrifice is mandatory (permanent neurological deficit expected)
- Functional consequences (e.g., foot drop if sciatic nerve, hand weakness if median/ulnar)
- Alternative of amputation (better local control 95% vs 80% limb salvage, same survival)
- Need for adjuvant radiotherapy
- Recurrence and metastasis risk (30-50% local, 40-65% distant)
Realistic expectations are essential given poor prognosis.
Complications
Treatment-Related Complications
- Incidence
- 20-30%
- Risk Factors
- Preoperative radiation (30-40%), large resection, poor nutrition
- Management
- VAC therapy, debridement, flap coverage
- Incidence
- 40-60%
- Risk Factors
- Expected with nerve resection (informed consent critical)
- Management
- Physiotherapy, tendon transfers, orthotics, occupational therapy
- Incidence
- 5-10%
- Risk Factors
- Vessel encasement, difficult dissection
- Management
- Vascular repair or reconstruction, anticoagulation
- Incidence
- Variable
- Risk Factors
- Postoperative radiation, high dose
- Management
- Physiotherapy, surgical release if severe
Disease-Related Complications
- Incidence: 30-50% at 5 years despite wide resection
- Risk factors: Positive margins, large size (greater than 10 cm), trunk location, high grade
- Median time to recurrence: 12-24 months (80% within 2 years)
- Management: Re-resection if feasible, radiotherapy if not previously given, amputation, palliative care
- Incidence: 40-65% at 5 years
- Most common site: Lung (80-90% of metastases)
- Other sites: Bone, liver, brain (rare)
- Median time to metastasis: 18 months
- Management: Palliative chemotherapy, metastasectomy (selected cases), best supportive care
- Indications: Limited lung metastases (1-3 nodules), disease-free interval greater than 12 months, complete resection possible, good performance status
- Improves survival in selected patients: 5-year survival 30-40% versus 10% with chemotherapy alone
Postoperative Care and Surveillance
Immediate Postoperative Period
Postoperative Management
- Wound monitoring (flap viability, drainage volume)
- DVT prophylaxis (mechanical plus pharmacological)
- Pain management (multimodal analgesia, avoid opioid dependence)
- Assess neurological deficit (document expected vs unexpected)
- Drain management (remove when less than 30 mL per 24 hours)
- Wound assessment (infection, dehiscence, hematoma)
- Early mobilization with physiotherapy
- Occupational therapy for functional adaptation
- Wound check, suture removal
- Final histology review (margin status, grade)
- MDT discussion of adjuvant therapy
- Radiation oncology consultation if indicated
If radiotherapy indicated:
- Start 6-8 weeks post-op (wound healed)
- 60-66 Gy over 6-7 weeks
- Continue physiotherapy during treatment
Treatment completion marks start of surveillance phase.
Surveillance Protocol
- Clinical Exam
- Every 3 months
- Chest Imaging
- CT chest every 3 months
- MRI Primary Site
- Every 3-6 months
- Rationale
- 80% recurrence within 2 years
- Clinical Exam
- Every 4-6 months
- Chest Imaging
- CT chest every 4-6 months
- MRI Primary Site
- Every 6-12 months
- Rationale
- 90% recurrence within 5 years
- Clinical Exam
- Annually
- Chest Imaging
- CT chest annually
- MRI Primary Site
- As clinically indicated
- Rationale
- Late recurrence possible
Rationale:
- 80% of recurrences occur within 2 years (intensive early surveillance)
- 90% of recurrences occur within 5 years
- Lung is most common metastatic site (50%)
- Early detection may allow metastasectomy in selected cases
Outcomes and Prognosis
Survival
MPNST has the WORST prognosis among soft tissue sarcomas.
5-Year Survival by Etiology:
- 5-Year Survival
- 21%
- Median Age
- 26-30 years
- Key Features
- Plexiform transformation, younger, worse prognosis
- 5-Year Survival
- 42%
- Median Age
- 40-50 years
- Key Features
- De novo, better prognosis than NF1
- 5-Year Survival
- 20-30%
- Median Age
- Variable
- Key Features
- Latency 10-20 years, poor prognosis
Prognostic Factors
Favorable Prognostic Factors:
- Complete surgical resection (R0 margins)
- Small size (less than 5 cm)
- Superficial location (above fascia)
- Low grade (rare, under 10% of MPNST)
- Distal extremity location
- Sporadic (non-NF1)
Unfavorable Prognostic Factors:
R0 (negative margins): 65% 5-year survival
R1 (microscopic positive): 40% 5-year survival
R2 (gross residual): 30% 5-year survival
Margin status is single most important modifiable prognostic factor.
- Large size (greater than 10 cm)
- Deep location (subfascial)
- High grade (Grade 3, 70% of MPNST)
- Trunk or proximal extremity
- Presence of necrosis (greater than 50%)
- High mitotic count (greater than 20 per 10 HPF)
- NF1-associated (worse than sporadic)
- Radiation-induced
- Malignant Triton tumor variant
- Age greater than 50 years
- Inadequate initial surgery (marginal excision)
- Delay in diagnosis and treatment
- Inability to deliver adjuvant radiotherapy
- Poor response to chemotherapy
Guidelines, Registries & Global Practice
Global Epidemiology
MPNST is rare but disproportionately important because of its NF1 association and poor outcomes. According to PubMed-indexed evidence, the incidence in the general clinic population is approximately 0.001% versus 4.6% in neurofibromatosis (Ducatman et al., Cancer 1986, DOI), and the lifetime risk in NF1 is 8-13% (Evans et al., J Med Genet 2002, DOI). MPNST accounts for roughly 5-10% of all soft-tissue sarcomas. NF1-associated disease presents around two decades earlier (median 26 years) than sporadic disease (median 62 years), a pattern reproduced across European, North American and Asian series. Geographic incidence is broadly similar worldwide because NF1 prevalence (~1 in 3000 births) is consistent across populations; differences in reported outcomes largely reflect access to specialist sarcoma centres rather than true biological variation.
International Guideline Comparison
- Diagnostic Pathway
- MRI + core biopsy at sarcoma centre; consider FDG-PET in NF1
- Surgery
- Wide excision to negative margins; limb preservation when feasible
- Adjuvant Therapy
- Radiotherapy for high-grade / large / close margins; chemotherapy selective
- Evidence Level
- Category 2A (consensus)
- Diagnostic Pathway
- Refer to reference centre before biopsy; image-guided core biopsy
- Surgery
- Wide R0 excision as cornerstone; MDT planning mandatory
- Adjuvant Therapy
- Radiotherapy improves local control; chemotherapy not standard adjuvant
- Evidence Level
- Expert consensus, limited RCT data
- Diagnostic Pathway
- Mandatory sarcoma MDT; suspected sarcoma 2-week-wait referral
- Surgery
- Planned excision by sarcoma surgeon; avoid unplanned 'whoops' excision
- Adjuvant Therapy
- Adjuvant radiotherapy for high-grade STS; chemotherapy in trials
- Evidence Level
- Guideline (NICE sarcoma pathway)
- Diagnostic Pathway
- Standardised STS trial pathways
- Surgery
- R0 resection endpoint
- Adjuvant Therapy
- Doxorubicin-ifosfamide best first-line response in advanced disease
- Evidence Level
- Level 2 (pooled trial analysis)
There is broad international agreement on the core principles: referral to a specialist sarcoma centre before biopsy, image-guided core biopsy with the tract sited for en-bloc excision, wide R0 surgical resection as the only curative modality, and adjuvant radiotherapy to improve local (but not overall) control of high-grade tumours. The main area of practice variation is the role of chemotherapy, which is offered selectively (young patients, large high-grade tumours, or downstaging) without proven survival benefit; the EORTC pooled analysis shows advanced MPNST responds to chemotherapy comparably to other soft-tissue sarcomas (Kroep et al., Ann Oncol 2011, DOI).
Registry & Reference-Centre Evidence
- Outcomes are consistently better in high-volume sarcoma centres: contemporary multidisciplinary series report 5- and 10-year disease-specific survival of 60% and 45% (Stucky et al., Ann Surg Oncol 2012, DOI), an improvement over historical series.
- National soft-tissue sarcoma pathways (UK NICE/BSG, European EURACAN reference networks, and registry-linked centres such as the Australian sarcoma services) all mandate MDT review and discourage unplanned ("whoops") excisions, which worsen margins and survival.
- Molecular confirmation with H3K27me3 immunohistochemistry is now widely available and recommended in difficult cases (Prieto-Granada et al., Am J Surg Pathol 2016, DOI).
Specialist Referral Principle
Any suspected MPNST - particularly a rapidly enlarging or painful mass in an NF1 patient - should be referred to a specialist sarcoma multidisciplinary team before biopsy or excision. Unplanned excision of a presumed benign neurofibroma that proves to be MPNST compromises margins and is a leading cause of preventable poor outcomes worldwide. Examples of national sarcoma networks include the NHS specialist sarcoma centres (UK), EURACAN reference centres (Europe), NCCN-designated centres (USA) and the state-based Australian sarcoma services.
Medicolegal Considerations
- Pre-biopsy imaging and staging completed and reviewed
- Biopsy performed by or in consultation with sarcoma team (oncologically sound technique)
- MDT discussion documented before definitive surgery with treatment recommendation
- Informed consent including: poor prognosis (5-year survival 40-60%), nerve sacrifice requirement, neurological deficit expected, need for adjuvant therapy, recurrence and metastasis risk
- Excision of suspected neurofibroma without imaging/biopsy (missed MPNST, inadequate margins)
- Marginal or enucleation excision rather than wide 2 cm margins (high recurrence)
- Failure to refer to sarcoma MDT before treatment
- Inadequate surveillance leading to late detection of recurrence or metastasis
- Not discussing amputation as alternative when limb salvage results in positive margins
MCQ Practice Points
Q: What percentage of MPNST occur in NF1 patients, and what is the 5-year survival?
A: 50% of MPNST occur in NF1 patients, with 21% 5-year survival (versus 42% in sporadic MPNST). This is significantly worse prognosis. The lifetime risk of MPNST in NF1 patients is 8-13% overall, but 25-30% in those with plexiform neurofibromas. Median age at diagnosis is 26 years in NF1 versus 40-50 years in sporadic.
Q: What SUVmax threshold on PET-CT suggests MPNST in NF1 patients? What is the sensitivity and specificity?
A: FDG-PET diagnosed NF1-associated MPNST with 89% sensitivity and 95% specificity (Ferner et al., Annals of Oncology 2008). A SUVmax less than 2.5 is reassuring while SUVmax greater than 3.5 raises high suspicion; the 2.5-3.5 range is intermediate and warrants close surveillance or biopsy. Note that SUVmax does not reliably predict tumour grade.
Q: What are the key molecular alterations in MPNST pathogenesis?
A: NF1 loss plus TP53 inactivation (75%) plus PRC2 complex mutations (SUZ12 or EED in 70-90%). The progression is: germline NF1 mutation, somatic second hit causes neurofibroma, additional TP53 loss and PRC2 loss drives malignant transformation to MPNST. H3K27me3 loss (due to PRC2 mutation) is a diagnostic immunohistochemistry marker supporting MPNST.
Q: What surgical margin is required for MPNST and what is the impact of margin status on survival?
A: Wide excision with 2 cm margins en bloc with involved nerve. Margin status is the most important prognostic factor: R0 resection (negative margins) achieves 65% 5-year survival, R1 (microscopic positive) 40% survival, R2 (gross residual) 30% survival. Re-excision is mandatory if positive margins on final pathology.
Q: Does adjuvant radiotherapy improve survival in MPNST?
A: Radiotherapy improves LOCAL CONTROL from 60-70% to 80-85% but does NOT improve overall survival. Dose is 60-66 Gy postoperatively or 50 Gy preoperatively. Indications: high-grade MPNST, positive/close margins, large size greater than 5 cm, deep location. Chemotherapy has modest benefit - response rate 20-30% (lower than other sarcomas 40-50%), no proven survival benefit for adjuvant chemotherapy.
Q: What percentage of MPNST are S100 positive and what is the staining pattern?
A: Only 50-70% of MPNST are S100 positive, and staining is FOCAL and PATCHY (not diffuse). This is unlike schwannoma which is diffusely and strongly S100 positive. S100 negativity does NOT exclude MPNST. Other markers: SOX10 50-60% positive, H3K27me3 loss in 50-70% (PRC2 mutation indicator), Ki67 high (greater than 10%, often 30-50%).
Exam Viva Scenarios
Practise clinical reasoning and management decisions out loud
“A 28-year-old woman with NF1 presents with a rapidly enlarging painful mass in the proximal thigh that has grown over 3 months. She has a known large plexiform neurofibroma in this location that was stable for years. MRI shows a 9 cm heterogeneous mass with necrosis arising from the sciatic nerve. PET-CT shows SUV of 5.2 and no distant metastases. Core needle biopsy confirms high-grade MPNST. How would you manage this patient?”
“What is the role of PET-CT in the diagnosis and management of MPNST, particularly in NF1 patients? Discuss the evidence and clinical utility including SUV thresholds.”
“You performed wide excision of a 12 cm proximal arm MPNST in a 45-year-old man. Final pathology returns as high-grade MPNST with a positive deep margin - tumor extends to within 1 mm of the radial nerve which you preserved. All other margins are negative with 2 cm clearance. The patient is 14 days post-operative with healing wound. How do you proceed?”
Definition and Epidemiology
- Aggressive sarcoma from peripheral nerve sheath, 5-10% of all soft tissue sarcomas
- 50% NF1-associated (plexiform transformation 25-30% lifetime risk), 40% sporadic, 10% radiation-induced
- Median age: 26-30 years in NF1, 40-50 years in sporadic
- Location: 40-50% proximal extremity, 25-30% trunk, 15-20% head/neck
- WORST PROGNOSIS of all soft tissue sarcomas
Molecular Pathogenesis (MPNST Mnemonic)
- M - Malignant transformation: Plexiform neurofibroma to MPNST in NF1
- P - PRC2 mutations: SUZ12 or EED loss in 70-90%, H3K27me3 loss diagnostic
- N - NF1 association: 50% of MPNST, worse prognosis than sporadic
- S - S100 focal positive: Only 50-70% and FOCAL not diffuse
- T - TP53 mutations: 75% have TP53 inactivation
HIGH RISK Features (Mnemonic)
- H - Heterogeneous MRI, I - Infiltrative margins, G - Growth rapid
- H - Hard consistency (firm vs soft neurofibroma)
- R - Radiotherapy history (10% radiation-induced), I - Intense pain (60-70%)
- S - Size greater than 5 cm (mean 8-10 cm), K - Ki67 high (greater than 10%)
Investigations
- MRI local staging: Heterogeneous signal, necrosis, irregular margins, loss of target sign
- PET-CT: SUV less than 2.5 benign (NPV 100%), 2.5-3.5 intermediate, greater than 3.5 MPNST (89% sens, 95% spec)
- CT chest for lung metastases (most common site, 50% at presentation or follow-up)
- Core needle biopsy 14-16G: H&E, S100, SOX10, Ki67, H3K27me3 IHC
- AJCC staging: Most present Stage IIIB (large, deep, high-grade) or IV (metastatic)
Histopathology
- High-grade 90%: Spindle cells, high mitoses greater than 10/10 HPF, necrosis 50-70%
- S100 positive only 50-70%, FOCAL not diffuse (negative does NOT exclude)
- H3K27me3 loss in 50-70% (PRC2 mutation marker)
- FNCLCC grading: Grade 3 (high) in 70% at diagnosis
- Variants: Triton tumor (5-10%, rhabdomyoblastic, worse prognosis)
Surgical Management
- Wide excision 2 cm margins en bloc with involved nerve (nerve sacrifice mandatory)
- Biopsy tract excised en bloc
- R0 resection: 65% 5-year survival vs R1 40%, R2 30%
- Amputation if neurovascular bundle encased (95% local control vs 80% limb salvage, same survival)
- Limb salvage achievable in greater than 95% but significant neurological deficit
Adjuvant Therapy
- Radiotherapy 60-66 Gy postop or 50 Gy preop: Improves local control 60-70% to 80-85% but NOT survival
- Indications: High-grade, positive/close margins, greater than 5 cm, deep
- Chemotherapy: Modest benefit, response rate 20-30% (vs 40-50% other sarcomas)
- No proven survival benefit for adjuvant chemotherapy
- NF1-MPNST particularly resistant to chemotherapy
Prognosis and Surveillance
- 5-year survival: NF1-MPNST 21%, sporadic 42%, overall 40-60%, metastatic 10-15%
- Local recurrence 30-50%, distant metastasis 40-65% (lung 80-90% of mets)
- 80% recurrence within 2 years, 90% within 5 years
- Surveillance: Q3mo exam/CT/MRI years 1-2, Q6mo years 3-5, annual after 5 years
- Margin status most important prognostic factor (R0 vs R1/R2)
Evidence Base and Key Studies
Malignant Transformation Risk in NF1 (Landmark Population Study)
- Population-based longitudinal study, NW England (4.1 million, 1984-1996): 21 NF1-associated and 37 sporadic MPNST
- Lifetime risk of MPNST in NF1 estimated at 8-13% (much higher than prior cross-sectional 1-2%)
- Median age at MPNST diagnosis: 26 years in NF1 versus 62 years in sporadic disease (p less than 0.001)
- Five-year survival from diagnosis: 21% NF1-associated versus 42% sporadic (p=0.09)
- First study to establish true lifetime risk and justify low threshold for investigation in NF1
FDG-PET for Detecting Malignant Transformation in NF1
- Long-term clinical study: 116 lesions in 105 NF1 patients with symptomatic plexiform neurofibromas (29 MPNST confirmed)
- FDG PET and PET-CT diagnosed NF1-associated tumours with sensitivity 0.89 (95% CI 0.76-0.96) and specificity 0.95 (95% CI 0.88-0.98)
- No MPNST was diagnosed on clinical follow-up of 23 lesions called benign on PET (high negative predictive value)
- SUVmax level did NOT reliably predict tumour grade - other tracers needed for grading
- Recommended as a sensitive and specific diagnostic tool for NF1-associated MPNST
Surgical Outcomes and Prognostic Factors (Mayo Clinic)
- Retrospective review of 175 MPNST patients (1985-2010); median age 44 years, median tumour size 6 cm, 61% high-grade
- Surgical resection in 95%; margins R0 69%, R1 2%, R2 9%, unknown 20%; radiation given in 42%
- Five- and ten-year disease-specific survival 60% and 45%; local recurrence rate 22%
- Independent poor prognostic factors on multivariate analysis: size 5 cm or greater (HR 6.1), local recurrence (HR 4.4), high grade (HR 3.8), truncal location (HR 3.7)
- Outcomes better than historically reported, attributed to multidisciplinary management
First-Line Chemotherapy in MPNST (EORTC Pooled Analysis)
- Retrospective pooled analysis of 175 MPNST among 2675 STS patients across 12 EORTC Soft Tissue and Bone Sarcoma Group trials
- Outcomes were SIMILAR for MPNST versus other STS: response rate 21% versus 22%, median PFS 17 versus 16 weeks, overall survival 48 versus 51 weeks
- Doxorubicin-ifosfamide produced the best response; single-agent ifosfamide had the worst prognosis
- Performance status was an independent prognostic factor for overall survival
- Refutes the older belief that MPNST is intrinsically more chemoresistant than other soft-tissue sarcomas
PRC2 Inactivation in MPNST Pathogenesis (Defining Molecular Study)
- Loss-of-function alterations of PRC2 components (EED or SUZ12) found in 92% of sporadic, 70% of NF1-associated and 90% of radiotherapy-associated MPNST
- PRC2 loss causes complete loss of H3K27me3 and aberrant activation of PRC2-repressed homeobox master regulators
- CDKN2A altered in 81% and NF1 in 72% of non-NF1-associated MPNST, co-occurring with PRC2 loss
- Restoring the lost PRC2 component in a deficient cell line recovered H3K27me3 and reduced growth
- Establishes cooperative NF1 + CDKN2A + PRC2 loss as the core genomic signature of MPNST
Clinicopathologic Behaviour of MPNST (Classic Series)
- Foundational clinicopathologic study of 120 MPNST over a 71-year period; mean age 35 years
- 62 of 120 (52%) arose in neurofibromatosis and 13 (11%) were post-radiation sarcomas
- Incidence of MPNST in neurofibromatosis 4.6% versus 0.001% in the general clinic population
- Tumour greater than 5 cm and neurofibromatosis independently worsened prognosis (p less than 0.05)
- Survival improved with total rather than subtotal resection, most marked in small lesions
References
-
Ducatman BS, Scheithauer BW, Piepgras DG, et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57(10):2006-2021. PMID:3082508. doi:10.1002/1097-0142(19860515)57:10%3C2006::aid-cncr2820571022%3E3.0.co;2-6
-
Evans DGR, Baser ME, McGaughran J, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39(5):311-314. doi:10.1136/jmg.39.5.311
-
Lee W, Teckie S, Wiesner T, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46(11):1227-1232. doi:10.1038/ng.3095
-
Ferner RE, Golding JF, Smith M, et al. [18F]2-fluoro-2-deoxy-D-glucose positron emission tomography (FDG PET) as a diagnostic tool for neurofibromatosis 1 (NF1) associated malignant peripheral nerve sheath tumours (MPNSTs): a long-term clinical study. Ann Oncol. 2008;19(2):390-394. doi:10.1093/annonc/mdm450
-
Prieto-Granada CN, Wiesner T, Messina JL, et al. Loss of H3K27me3 expression is a highly sensitive marker for sporadic and radiation-induced MPNST. Am J Surg Pathol. 2016;40(4):479-489. doi:10.1097/PAS.0000000000000564
-
Stucky CC, Johnson KN, Gray RJ, et al. Malignant peripheral nerve sheath tumors (MPNST): the Mayo Clinic experience. Ann Surg Oncol. 2012;19(3):878-885. doi:10.1245/s10434-011-1978-7
-
Kroep JR, Ouali M, Gelderblom H, et al. First-line chemotherapy for malignant peripheral nerve sheath tumor (MPNST) versus other histological soft tissue sarcoma subtypes and as a prognostic factor for MPNST: an EORTC soft tissue and bone sarcoma group study. Ann Oncol. 2011;22(1):207-214. doi:10.1093/annonc/mdq338
-
Anghileri M, Miceli R, Fiore M, et al. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer. 2006;107(5):1065-1074. doi:10.1002/cncr.22098
-
Wasa J, Nishida Y, Tsukushi S, et al. MRI features in the differentiation of malignant peripheral nerve sheath tumors and neurofibromas. AJR Am J Roentgenol. 2010;194(6):1568-1574.
-
National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Soft Tissue Sarcoma Version 2.2024.