ECM Macromolecules | 28 Collagen Types | GAG Side Chains | Triple Helix Structure
- Collagen is most abundant protein in mammals (25-35% of total body protein)
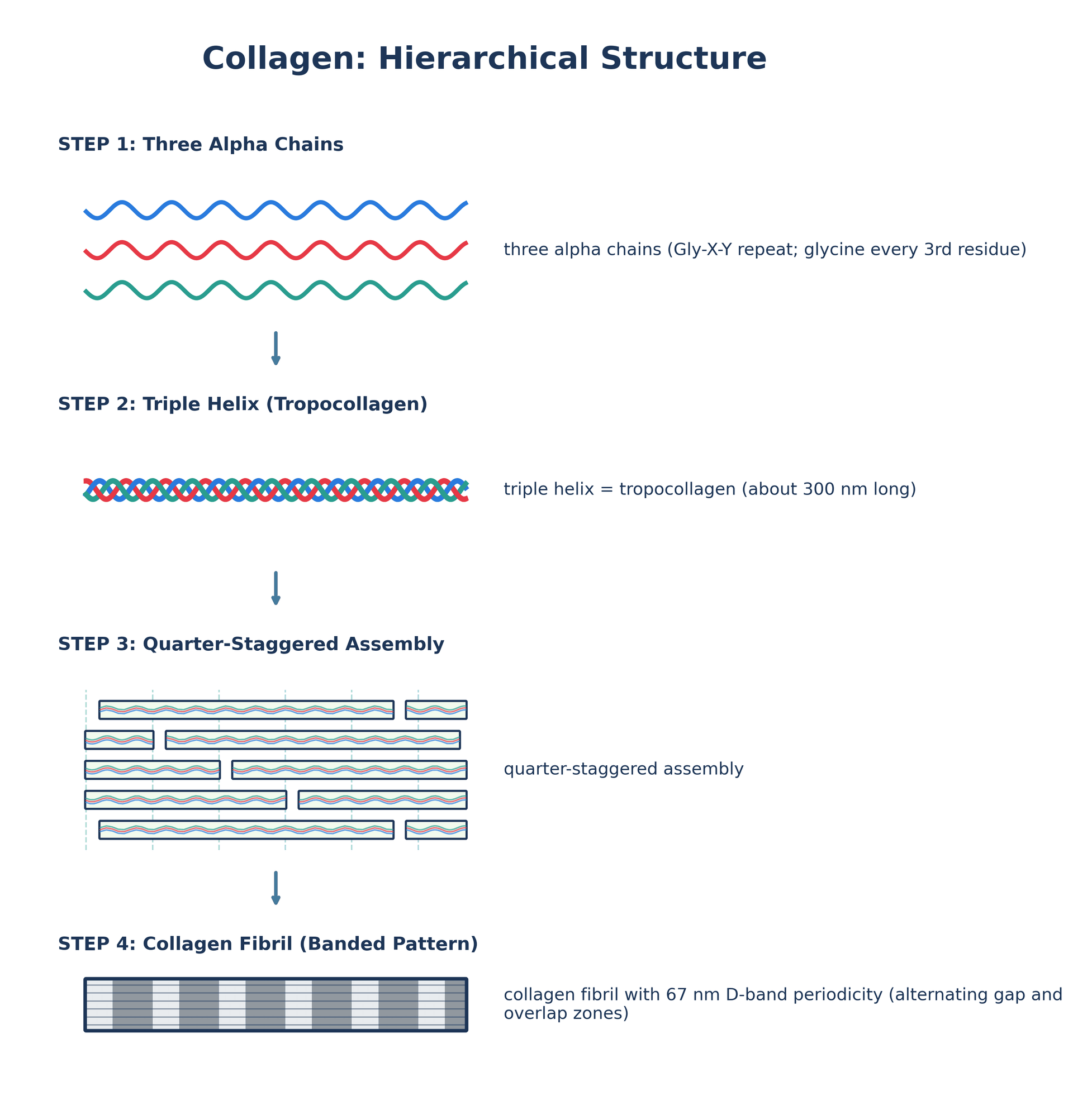
- Triple helix structure - Gly-X-Y repeat with glycine every third residue
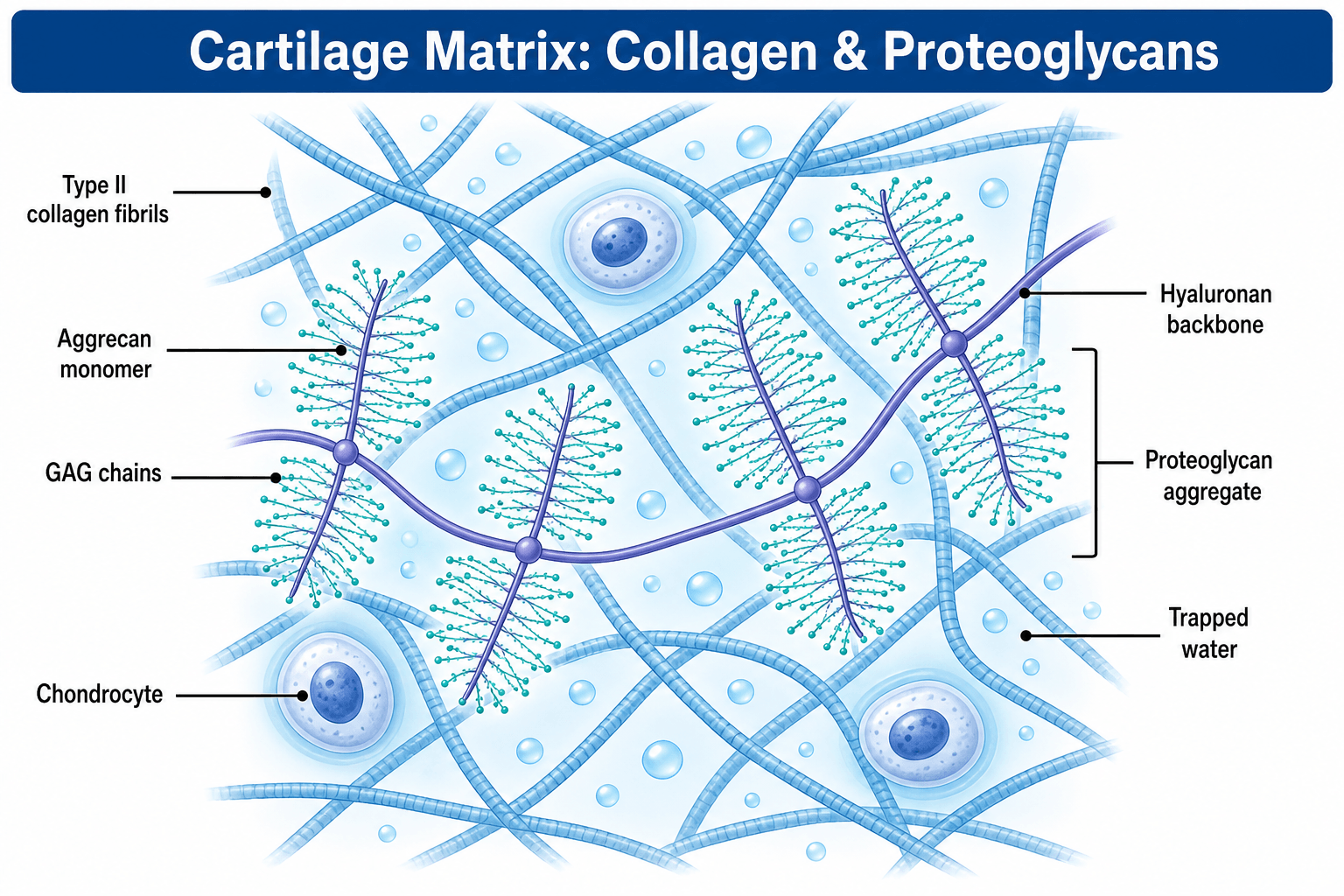
- Proteoglycans consist of core protein with GAG side chains
- Aggrecan is major cartilage proteoglycan, decorin binds collagen
- Hydroxylation requires Vitamin C - scurvy impairs collagen synthesis
- “Type I collagen mutations cause osteogenesis imperfecta
- “Type II collagen mutations cause chondrodysplasias
- “Lysyl oxidase creates crosslinks (requires copper)
- “GAG side chains on proteoglycans attract water via negative charge
Collagen has unique triple helix with Gly-X-Y amino acid repeat. Glycine (smallest amino acid) must be every third residue to fit in center of helix. Proline and hydroxyproline common in X and Y positions. Mutations replacing glycine cause structural diseases (OI, EDS).
Hydroxylation of proline and lysine requires Vitamin C (cofactor for prolyl and lysyl hydroxylases). Scurvy causes defective collagen. Lysyl oxidase (requires copper) creates crosslinks between collagen molecules, essential for tensile strength.
Proteoglycans are core protein with GAG side chains (chondroitin sulfate, keratan sulfate, heparan sulfate, dermatan sulfate). GAGs are repeating disaccharides with sulfate/carboxyl groups creating negative charge. Attract water and cations.
Type I collagen: bone (90%), tendon, ligament, skin. Type II: hyaline cartilage, nucleus pulposus. Type III: blood vessels, healing wounds (with Type I). Type IV: basement membranes. Know which type in which tissue.
Overview
Collagen and proteoglycans are major components of extracellular matrix in connective tissues including bone, cartilage, tendon, ligament, and skin.
Why collagen and proteoglycans matter clinically:
Mutations in collagen genes cause osteogenesis imperfecta (Type I), chondrodysplasias (Type II), vascular Ehlers-Danlos syndrome (Type III), and Alport syndrome (Type IV). Understanding collagen structure explains disease mechanisms.
Collagen synthesis is essential for fracture healing, tendon repair, and wound closure. Type III collagen appears early in healing, later replaced by Type I. Factors impairing collagen synthesis (vitamin C deficiency, medications) delay healing.
Osteoarthritis involves proteoglycan loss and collagen network disruption. Intervertebral disc degeneration involves aggrecan degradation. Understanding normal structure is essential to understand pathology.
Collagen-based scaffolds are used in cartilage repair and tissue engineering. Understanding collagen structure, crosslinking, and degradation informs biomaterial design.
Collagen is the most abundant protein in mammals (25-35% of total body protein). It provides structural framework and tensile strength in connective tissues. The unique triple helix structure with Gly-X-Y repeat is essential for collagen function.
Collagen and proteoglycans are completed by elastin, the structural protein responsible for elastic recoil in tissues that must stretch and rebound — the ligamentum flavum (the most elastin-rich ligament, roughly 80% elastin, which is why it stays taut and does not buckle into the spinal canal), aorta, skin and lung. Soluble tropoelastin is secreted and deposited onto a scaffold of fibrillin-1 microfibrils, then crosslinked by lysyl oxidase (the same copper-dependent enzyme used for collagen) into the insoluble polymer via the unique desmosine and isodesmosine crosslinks. Clinically: fibrillin-1 (FBN1) mutations cause Marfan syndrome (the microfibril defect dysregulates TGF-beta signalling), elastin (ELN) defects cause cutis laxa and supravalvular aortic stenosis (Williams syndrome), and copper-dependent crosslinking failure (Menkes, lathyrism) weakens elastin as well as collagen.
Concepts and Molecular Structure
Collagen Triple Helix
Primary structure - Gly-X-Y repeat:
- Glycine every third residue (Gly-X-Y)
- X position: Often proline (28%)
- Y position: Often hydroxyproline (38%)
- Glycine is smallest amino acid, fits in helix center
- Any other amino acid at glycine position disrupts helix
Secondary structure - Left-handed helix:
- Each alpha chain forms left-handed helix (polyproline II type)
- 3 residues per turn
- Extended structure (not compact like alpha-helix)
Tertiary structure - Right-handed superhelix:
- Three alpha chains wrap around each other
- Right-handed triple helix
- Glycine residues at central axis (every third residue)
- Proline and hydroxyproline stabilize helix
- No internal hydrogen bonds within chains
- Hydrogen bonds between chains
Tropocollagen molecule:
- 300 nm length
- 1.5 nm diameter
- Molecular weight: ~300 kDa
- Three chains: Type I has two alpha-1(I) and one alpha-2(I)
- Type II has three identical alpha-1(II) chains
Glycine substitutions in Type I collagen cause osteogenesis imperfecta. Glycine is the only amino acid small enough to fit in the center of the triple helix. Replacing glycine with larger amino acids (e.g., serine, cysteine) disrupts helix structure, causing brittle bones. Location of mutation affects severity.
The triple helix structure is fundamental to collagen function and stability.
Major Collagen Types
28 collagen types have been identified. The major types relevant to orthopaedics are Types I, II, III, IV, V, IX, X, and XI.
- Structure
- Fibril-forming [α1(I)]₂α2(I)
- Tissue Distribution
- Bone, tendon, ligament, skin, dentin
- Function
- Tensile strength, structural support
- Clinical Significance
- OI: mutations in COL1A1, COL1A2
- Structure
- Fibril-forming [α1(II)]₃
- Tissue Distribution
- Hyaline cartilage, vitreous humor, nucleus pulposus
- Function
- Compression resistance in cartilage
- Clinical Significance
- Chondrodysplasias, early OA
- Structure
- Fibril-forming [α1(III)]₃
- Tissue Distribution
- Blood vessels, skin, reticular fibers, healing tissue
- Function
- Elastic recoil, early wound healing
- Clinical Significance
- Vascular EDS (Type IV EDS)
- Structure
- Network-forming [α1(IV)]₂α2(IV)
- Tissue Distribution
- Basement membranes (all)
- Function
- Filtration barrier, cell attachment
- Clinical Significance
- Alport syndrome (kidney, ear, eye)
- Structure
- Fibril-forming [α1(V)]₂α2(V)
- Tissue Distribution
- Bone, cornea, with Type I
- Function
- Regulates Type I fibril diameter
- Clinical Significance
- Classical EDS (with Type I)
- Structure
- FACIT [α1(IX)]α2(IX)α3(IX)
- Tissue Distribution
- Cartilage, vitreous, with Type II
- Function
- Links Type II fibrils, resists shear
- Clinical Significance
- Multiple epiphyseal dysplasia
- Structure
- Network-forming [α1(X)]₃
- Tissue Distribution
- Hypertrophic cartilage (growth plate)
- Function
- Endochondral ossification
- Clinical Significance
- Schmid metaphyseal chondrodysplasia
- Structure
- Fibril-forming [α1(XI)]α2(XI)α3(XI)
- Tissue Distribution
- Cartilage, vitreous, with Type II
- Function
- Regulates Type II fibril diameter
- Clinical Significance
- Stickler syndrome (with Type II)
Type I collagen (bone, tendon, ligament) forms larger diameter fibrils (50-200 nm) and has high tensile strength. Type II collagen (cartilage) forms smaller diameter fibrils (20-40 nm) optimized for compression resistance. Type I has two alpha-1(I) and one alpha-2(I) chain. Type II has three identical alpha-1(II) chains (homotrimer).
Collagen classification:
- Fibril-forming: Types I, II, III, V, XI (form D-banded fibrils)
- FACIT (Fibril-Associated Collagens with Interrupted Triple helices): Types IX, XII, XIV
- Network-forming: Types IV, VIII, X (basement membranes, specialized networks)
- Anchoring fibrils: Type VII (epidermis-dermis junction)
- Transmembrane: Types XIII, XVII, XXIII
Understanding tissue-specific collagen types is essential for orthopaedic basic science.
Differential Diagnosis: Heritable Collagen/ECM Disorders
Glycine substitutions, defective hydroxylation/crosslinking, and proteoglycan-processing defects all converge on connective-tissue fragility but differ in the molecule affected and the dominant phenotype. This table is high-yield for distinguishing the classic ECM diseases in a viva.
- Molecule / Gene
- Type I collagen (COL1A1/COL1A2)
- Defect Mechanism
- Glycine substitution or haploinsufficiency
- Key Clinical Clue
- Fragile bones, blue sclerae, dentinogenesis imperfecta
- Molecule / Gene
- Type III collagen (COL3A1)
- Defect Mechanism
- Reduced/abnormal type III collagen
- Key Clinical Clue
- Arterial, bowel and uterine rupture; thin translucent skin
- Molecule / Gene
- Type V collagen (COL5A1/COL5A2)
- Defect Mechanism
- Disordered type I fibril regulation
- Key Clinical Clue
- Skin hyperextensibility, atrophic scars, hypermobility
- Molecule / Gene
- Type II collagen (COL2A1)
- Defect Mechanism
- Defective cartilage collagen
- Key Clinical Clue
- Short stature, myopia/retinal detachment, early OA
- Molecule / Gene
- Type IV collagen (COL4A3-5)
- Defect Mechanism
- Defective basement-membrane network
- Key Clinical Clue
- Haematuria/renal failure, sensorineural deafness
- Molecule / Gene
- Hydroxyproline (acquired)
- Defect Mechanism
- Vitamin C deficiency → no prolyl hydroxylation
- Key Clinical Clue
- Perifollicular haemorrhage, gum bleeding, poor healing
- Molecule / Gene
- Lysyl oxidase activity (ATP7A, copper)
- Defect Mechanism
- Copper deficiency → no crosslinking
- Key Clinical Clue
- Kinky hair, vascular tortuosity, bone fragility
- Molecule / Gene
- GAG-degrading enzymes (e.g. IDUA)
- Defect Mechanism
- Lysosomal GAG accumulation
- Key Clinical Clue
- Dysostosis multiplex, stiff joints, organomegaly
Clinical Relevance and Applications
Proteoglycans consist of core protein with covalently attached GAG side chains.
Proteoglycan Structure
Components:
- Core protein: Synthesized on ribosomes, varies in size
- GAG chains: Synthesized in Golgi, attached to core protein
- Link protein: Stabilizes aggrecan-hyaluronan binding (aggrecan only)
GAG attachment:
- GAGs attached to serine residues on core protein
- Tetrasaccharide linker: xylose-galactose-galactose-glucuronic acid
- Except keratan sulfate (attached to serine or threonine via N-acetylgalactosamine)
Size range:
- Small: Decorin (40 kDa core, 1 GAG chain, total 90-140 kDa)
- Medium: Perlecan (470 kDa core, multiple HS chains)
- Large: Aggrecan (220 kDa core, ~100 CS chains, total 2-3 million Da)
Proteoglycans vary in size and GAG composition depending on tissue function.
Distinct from the large aggregating aggrecan, lubricin (proteoglycan-4, PRG4, also called superficial zone protein/SZP) is a mucinous proteoglycan secreted by superficial-zone chondrocytes and synovial lining cells. Its central mucin domain carries O-linked GAG-like sugars that make it amphiphilic, so it adsorbs to the cartilage surface and provides boundary lubrication — the dominant low-velocity, high-load lubrication mode that protects the surface where a fluid film cannot form (working alongside hyaluronan and interstitial fluid pressurisation). Loss-of-function PRG4 mutations cause camptodactyly-arthropathy-coxa vara-pericarditis (CACP) syndrome (congenital joint contractures and non-inflammatory arthropathy), and lubricin depletion is implicated in post-traumatic and osteoarthritic surface wear.
Guidelines, Registries & Global Practice
Global Relevance and Epidemiology
- Collagen and proteoglycan biochemistry is core basic-science content across all major fellowship exams (FRCS Tr&Orth, FRACS, EBOT/FEBOT, ABOS, DNB/MS, MRCS, SICOT).
- Osteoarthritis, the dominant downstream disease of proteoglycan/collagen failure, affects an estimated 500+ million people worldwide and is a leading global cause of years lived with disability.
- Heritable disorders provide the natural "knockout" experiments: OI incidence is roughly 1 in 15,000-20,000 births; EDS collectively around 1 in 5,000.
Side-by-Side Guidance (ECM-Targeted OA Therapies)
- Glucosamine & Chondroitin
- Not recommended (strong, against)
- Intra-articular Hyaluronic Acid
- Not recommended for routine knee OA
- Glucosamine & Chondroitin
- Do not offer for OA
- Intra-articular Hyaluronic Acid
- Do not offer for OA management
- Glucosamine & Chondroitin
- Not recommended for disease modification
- Intra-articular Hyaluronic Acid
- Conditional/uncertain; context-dependent
- Glucosamine & Chondroitin
- Prescription crystalline glucosamine sulfate viewed more favourably
- Intra-articular Hyaluronic Acid
- May be considered as add-on in some algorithms
Registry & Practice Variation
- Joint registries (NJR UK, AJRR US, AOANJRR Australia, Swedish/Norwegian registries) capture the end-stage arthroplasty outcomes of cartilage matrix failure and increasingly track cartilage-repair and biologic procedures.
- High-resource settings: access to cell-based cartilage repair (ACI/MACI), osteochondral grafting and quantitative MRI of cartilage matrix (T1rho, dGEMRIC) for early matrix assessment.
- Limited-resource settings: emphasis on conservative load management, weight optimisation and addressing nutritional collagen cofactors (vitamin C, copper) where deficiency is endemic; advanced biologics are often unavailable.
Controversies & Areas of Uncertainty
Glasson's mouse data established ADAMTS5 as the dominant aggrecanase in rodents, but the relative contribution of ADAMTS4 versus ADAMTS5 in human disease remains debated, complicating translation of selective inhibitors.
GAG-precursor supplements are widely consumed, yet high-quality trials and meta-analyses show inconsistent, at best small, effects on pain and no convincing structure modification. Major guidelines (e.g. OARSI, NICE) do not recommend them for disease modification.
Despite a coherent biological rationale (restoring synovial HA), pooled evidence for clinically meaningful benefit in knee OA is weak and contested; recommendations differ markedly between societies.
Beyond collagen volume, the ratio of enzymatic (pyridinoline) to non-enzymatic advanced-glycation-end-product crosslinks is increasingly implicated in age- and diabetes-related bone fragility, but is not yet a routine clinical metric.
MCQ Practice Points
Q: What is the predominant proteoglycan in articular cartilage and its function?
A: Aggrecan is the major proteoglycan, attached to hyaluronic acid via link protein forming large aggregates. Contains glycosaminoglycan (GAG) side chains (chondroitin sulfate, keratan sulfate). Highly negatively charged, attracting water creating osmotic swelling pressure that resists compressive loads. Loss of aggrecan is early OA feature.
Q: What is the distribution of collagen types in articular cartilage?
A: Type II collagen: 90-95% of cartilage collagen, provides tensile strength. Type IX: Cross-links Type II fibrils. Type XI: Regulates fibril diameter. Type VI: Pericellular matrix around chondrocytes. Fibrocartilage (meniscus, labrum) contains Type I collagen. OA involves shift from Type II to Type I.
Q: What is the water content of articular cartilage and its significance?
A: Articular cartilage is 65-80% water by weight. Water content highest in superficial zone, lowest in deep zone. Creates biphasic viscoelastic behavior - fluid pressurization under load. Water bound to proteoglycans (fixed charge density). Dehydration decreases compressive stiffness. OA shows increased water content paradoxically.
Q: What are the structural zones of articular cartilage?
A: Superficial zone (10-20%): Collagen parallel to surface, highest water, flattened chondrocytes, lubricin secretion. Middle/transitional zone (40-60%): Random collagen orientation. Deep zone (30%): Collagen perpendicular, highest proteoglycan, columns of chondrocytes. Calcified zone: Anchors to subchondral bone via tidemark.
Q: What is the triple helix structure of collagen?
A: Three polypeptide chains (α-chains) wind into right-handed triple helix. Stabilized by glycine at every third position (smallest amino acid fits helix center). Proline and hydroxyproline provide rigidity. Hydroxyproline requires Vitamin C (scurvy causes collagen defects). Cross-linking between molecules provides tensile strength.
At a Glance
Collagen is the most abundant protein in mammals (25-35% of total body protein), characterised by a unique triple helix structure with the Gly-X-Y amino acid repeat. Type I collagen predominates in bone/tendon (90%), while Type II is found in hyaline cartilage. Proteoglycans consist of core proteins with negatively charged glycosaminoglycan (GAG) side chains that attract water - aggrecan is the major cartilage proteoglycan. Key exam points: Vitamin C is essential for proline/lysine hydroxylation (scurvy causes defective collagen), and lysyl oxidase (copper-dependent) creates crosslinks essential for tensile strength.
COLLAGENCOLLAGEN - Key Features
Hook:COLLAGEN is the common protein with glycine repeats forming triple helix
TYPESTYPES - Major Collagen Types
Hook:Know the TYPES of collagen: I in bone/tendon, II in cartilage, III in vessels, IV in basement membranes
PROTEOGLYCANSPROTEOGLYCANS - Structure and Function
Hook:PROTEOGLYCANS have protein core with negatively-charged GAG chains attracting water
Basic Science Viva Scenarios
Practise clinical reasoning and management decisions out loud
“Describe the structure of the collagen triple helix. What is the significance of the Gly-X-Y repeat?”
“Describe the steps of collagen biosynthesis from translation to fibril formation. Where are crosslinks formed and why are they important?”
“Describe the structure of aggrecan. How does it provide compressive stiffness in articular cartilage?”
Collagen Triple Helix
- Gly-X-Y repeat: glycine every 3rd residue (small enough for helix center)
- X = proline (28%), Y = hydroxyproline (38%)
- Three alpha chains: right-handed superhelix, 300 nm length, 1.5 nm diameter
- Type I: [α1(I)]₂α2(I), Type II: [α1(II)]₃ homotrimer
Collagen Biosynthesis
- Hydroxylation: Prolyl/lysyl hydroxylase (requires Vitamin C cofactor)
- Triple helix: C-terminal propeptides initiate assembly
- Propeptide cleavage: Procollagen → tropocollagen (300 nm)
- Fibril assembly: Quarter-stagger creates 67 nm D-band
- Crosslinks: Lysyl oxidase (requires copper) creates pyridinoline, deoxypyridinoline
Major Collagen Types
- Type I: Bone, tendon, ligament (90% of body collagen), OI mutations
- Type II: Hyaline cartilage, nucleus pulposus, chondrodysplasia mutations
- Type III: Blood vessels, skin, healing tissue, vascular EDS
- Type IV: Basement membranes (network-forming), Alport syndrome
Proteoglycan Structure
- Core protein + GAG side chains (chondroitin sulfate, keratan sulfate, etc)
- Aggrecan: 2-3 MDa, ~100 CS + ~60 KS chains
- Aggregates: 50-100 aggrecans bind hyaluronan via link protein (40-45 kDa)
- Decorin: Small (90-140 kDa), binds collagen, regulates fibril diameter
GAG Types
- Chondroitin sulfate (CS): GlcUA-GalNAc, 4- or 6-sulfate, cartilage/bone
- Keratan sulfate (KS): Gal-GlcNAc, 6-sulfate, cartilage (increases with age)
- Dermatan sulfate (DS): IdoUA-GalNAc, decorin GAG, regulates collagen
- Heparan sulfate (HS): High sulfation, basement membranes, growth factor binding
- Hyaluronan (HA): No sulfate, not protein-bound, aggregation backbone
Proteoglycan Function
- Fixed negative charge (SO₄⁻, COO⁻) attracts water via Donnan equilibrium
- Swelling pressure (0.1-0.3 MPa) provides compression resistance
- Collagen network constrains proteoglycan swelling (prestress)
- Aggrecan in cartilage: highest in deep zone (50-60 mg/mL)
Clinical Correlations
- Scurvy: Vitamin C deficiency → no hydroxyproline → unstable collagen
- Lathyrism: Lysyl oxidase inhibition (or Cu deficiency) → no crosslinks
- OI: Type I collagen mutations (glycine substitutions) → brittle bones
- OA: Aggrecanase (ADAMTS-4,5) cleaves aggrecan IGD → proteoglycan loss
- Urinary PYD/DPD: Bone resorption markers (crosslinks released)
Evidence Base
Collagen Structure and Stability
- Collagen is a right-handed bundle of three left-handed polyproline II-type helices
- Stereoelectronic effects and preorganisation (not just hydrogen bonding) drive triple-helix stability
- Hydroxyproline in the Y position stabilises the helix via inductive/stereoelectronic effects
- Self-assembly of type I tropocollagen reproduces native fibril properties in synthetic systems
Proteoglycan Form and Function: A Comprehensive Nomenclature
- Defines 43 distinct proteoglycan-encoding genes grouped into 4 classes by location
- Classes: intracellular, cell-surface, pericellular and extracellular proteoglycans
- Aggrecan (extracellular, modular) provides compressive stiffness in cartilage
- Small leucine-rich proteoglycans (decorin, biglycan) regulate collagen fibrillogenesis and signalling
Osteogenesis Imperfecta (Disease Primer)
- ~85% of OI is caused by dominant mutations in the type I collagen genes COL1A1 or COL1A2
- Mutations affect collagen quantity (haploinsufficiency, milder) or structure (glycine substitutions, more severe)
- Recessive, dominant and X-linked defects in collagen processing/modification and osteoblast genes cause the remaining cases
- Extra-skeletal features include blue sclerae, dentinogenesis imperfecta, hearing loss and cardiovascular involvement
The Role of Aggrecan in Normal and Osteoarthritic Cartilage
- Aggrecan bears chondroitin sulfate and keratan sulfate chains and aggregates with hyaluronan via link protein
- Both aggrecanases (ADAMTS) and matrix metalloproteinases cleave aggrecan, but at distinct sites
- Inflammation and overloading upregulate aggrecanolytic enzymes, depleting aggrecan in OA
- Synovial-fluid aggrecan fragments serve as a marker of ongoing cartilage destruction
Deletion of Active ADAMTS5 Prevents Cartilage Degradation (Landmark)
- First single-gene deletion shown to abrogate cartilage destruction in a model of OA
- Mice lacking the ADAMTS5 catalytic domain were protected after surgical joint destabilisation
- Identified ADAMTS5 (aggrecanase-2) as the primary aggrecanase in murine OA
- Established the aggrecanase IGD cleavage site as a rational therapeutic target
The 2017 International Classification of the Ehlers-Danlos Syndromes
- Replaced the 1998 Villefranche six-type nosology with 13 recognised EDS subtypes
- Most subtypes arise from defects in collagen genes or collagen-modifying enzymes
- Molecular confirmation is required for all subtypes except hypermobile EDS (clinical diagnosis)
- Vascular EDS (type III collagen / COL3A1) carries the highest risk of arterial and visceral rupture