Benign Peripheral Nerve Sheath Tumor | Eccentric Growth | Enucleation Preserves Nerve Function
- Eccentric growth displaces but does not infiltrate nerve fascicles (unlike neurofibroma)
- Antoni A (cellular, Verocay bodies) and Antoni B (hypocellular, myxoid) histological areas
- Target sign on MRI: peripheral T2 hyperintensity with central hypointensity (50-60% sensitive)
- Microsurgical enucleation preserves nerve function in 95% of cases
- NF2 (chromosome 22q12) causes bilateral vestibular schwannomas (pathognomonic)
- “Tinel sign positive: percussion over tumor elicits paresthesias in nerve distribution
- “Mobile perpendicular to nerve, fixed parallel to nerve (pathognomonic sign)
- “S100 protein strongly and diffusely positive on immunohistochemistry (unlike patchy in neurofibroma)
- “Ancient schwannoma shows degenerative atypia but remains benign (do not confuse with sarcoma)
- “Malignant transformation extremely rare (less than 1%) - malignant peripheral nerve sheath tumor arises de novo or from neurofibroma in NF1

CRITICAL DISTINCTION: Schwannoma is eccentric and encapsulated (can enucleate), neurofibroma infiltrates fascicles (cannot enucleate without nerve sacrifice). S100 diffuse strong in schwannoma, patchy in neurofibroma.
NF2 (chromosome 22q12): Bilateral vestibular schwannomas pathognomonic, multiple peripheral schwannomas. NF1: Neurofibromas (not schwannomas), cafe-au-lait spots, plexiform type transforms to MPNST.
Antoni A (cellular, palisading nuclei, Verocay bodies) and Antoni B (hypocellular, myxoid stroma). Ancient schwannoma shows degenerative atypia but is benign. S100 diffuse positive, encapsulated.
Microsurgical technique: Longitudinal epineurial incision, identify capsule, dissect tumor from displaced fascicles using nerve stimulator. Preserve nerve function in 95%. Recurrence less than 5% if complete.
SCHWANNSchwannoma Key Features
Hook:SCHWANN cells make schwannomas - remember the encapsulated eccentric growth that allows enucleation!
ECCENTRICSchwannoma vs Neurofibroma Distinction
Hook:ECCENTRIC growth is the key - schwannomas are eccentric to the nerve and can be enucleated!
Overview and Epidemiology
Schwannomas (also called neurilemmomas or neurinomas) are benign peripheral nerve sheath tumors arising from Schwann cells. Unlike neurofibromas which infiltrate nerve fascicles, schwannomas grow eccentrically and displace fascicles, creating a natural cleavage plane that allows microsurgical enucleation while preserving nerve function in 95% of cases. Schwannomas account for approximately 5% of all benign soft tissue tumors and are the most common tumor of peripheral nerves after neurofibromas.
The critical distinction between schwannoma and neurofibroma determines surgical approach: schwannomas can be enucleated preserving nerve function, while neurofibromas infiltrate fascicles and usually require nerve sacrifice for complete excision. Preoperative differentiation guides patient counseling about neurological outcomes.
Epidemiology
- Overall incidence: 1-2 per 100,000 population per year
- Peripheral nerve schwannomas: 90% solitary sporadic
- Vestibular schwannomas: 1 per 100,000 per year (most common cerebellopontine angle tumor)
- NF2-associated: Bilateral vestibular schwannomas in nearly 100% of NF2 patients by age 30
- Upper extremity: 40% (median, ulnar, radial nerves common)
- Lower extremity: 30% (sciatic, tibial, peroneal nerves)
- Head and neck: 25% (vestibular, vagus, sympathetic chain)
- Trunk: 5% (intercostal, paraspinal nerves)
- Vestibular schwannoma (acoustic neuroma): 8th cranial nerve, presents with unilateral hearing loss, tinnitus, imbalance
- Spinal schwannomas: Intradural extramedullary (most common), cause radiculopathy or myelopathy
- Mediastinal/retroperitoneal: Rare, often large at presentation
Pathophysiology and Anatomy
Cellular Origin and Pathogenesis
Schwannomas arise from Schwann cells, the myelinating cells of peripheral nerves. The tumor grows eccentrically from the nerve, surrounded by a well-defined capsule composed of epineurium and perineurium. Normal nerve fascicles are displaced to the periphery of the tumor rather than infiltrated, creating the surgical plane for enucleation.
- Origin: Neoplastic transformation of Schwann cells
- NF2 gene loss: Chromosome 22q12 (merlin/schwannomin)
- Merlin function: Links cell membrane to cytoskeleton, regulates cell growth
- Loss of heterozygosity: Two-hit hypothesis (germline plus somatic mutation in NF2)
- Eccentric growth: Displaces nerve fascicles peripherally
- Encapsulated: True capsule of compressed perineurium and epineurium
- Cleavage plane: Natural plane between tumor and nerve allows enucleation
- Slow growth: Mean growth rate 1-2 mm/year for vestibular schwannomas
Relationship to Nerve Fascicles
The fundamental difference between schwannoma and neurofibroma:
- Schwannoma
- Eccentric, displaces fascicles
- Neurofibroma
- Infiltrates within fascicles
- Schwannoma
- Well-encapsulated (compressed epineurium)
- Neurofibroma
- No true capsule
- Schwannoma
- Clear plane allows enucleation
- Neurofibroma
- No plane, fascicles run through tumor
- Schwannoma
- 95% preserve function with enucleation
- Neurofibroma
- Usually requires nerve sacrifice for excision
- Schwannoma
- Less than 5% if complete excision
- Neurofibroma
- 40-60% due to infiltrative nature
Neurofibromatosis Type 2 (NF2)
- NF2 gene located on chromosome 22q12
- Encodes merlin (schwannomin), a tumor suppressor protein
- Autosomal dominant inheritance with 50% penetrance by age 30, nearly 100% by age 60
- 50% sporadic (new mutation), 50% familial
- Bilateral vestibular schwannomas: Pathognomonic (nearly 100% of NF2 patients)
- Multiple schwannomas: Peripheral nerves, cranial nerves, spinal nerves
- CNS tumors: Meningiomas (45-58%), spinal ependymomas (33%)
- Cataracts: Juvenile posterior subcapsular or cortical (60-81%)
- Skin lesions: Fewer cafe-au-lait spots than NF1, schwannomas may be subcutaneous
Schwannomatosis - the Third Schwannoma Syndrome
The classic exam triad is NF1 versus NF2 versus schwannomatosis - and schwannomatosis is the one this topic must distinguish from NF2. Schwannomatosis is defined by multiple (usually non-vestibular) schwannomas in the ABSENCE of bilateral vestibular schwannomas - that absence is what separates it from NF2.
- Genetics: germline loss-of-function in SMARCB1 or LZTR1, both on chromosome 22 near the NF2 locus; many cases are sporadic.
- Dominant clinical feature: chronic, often disproportionate PAIN (rather than a hearing loss or a single mass), with peripheral and spinal schwannomas; intracranial meningiomas can occur (especially with SMARCB1). The cataracts and other NF2 stigmata are absent.
- Key caveat: a mosaic (segmental) NF2 can mimic schwannomatosis - molecular testing of the germline and of two separate tumours (searching for a shared NF2 variant) distinguishes them, as formalised in the updated international criteria.
- Management: pain control is central; excise symptomatic schwannomas; surveillance imaging; genetic counselling.
Exam point: multiple schwannomas plus chronic pain but NO bilateral vestibular schwannomas points to schwannomatosis (SMARCB1/LZTR1), not NF2 - and mosaic NF2 must be excluded genetically.
Histology and Pathology
Macroscopic Features
- Shape: Ovoid or fusiform mass along nerve
- Size: Typically 2-5 cm at presentation (can be larger)
- Capsule: Well-defined, smooth, glistening capsule
- Cut surface: Tan-yellow to grey-white, may show cystic degeneration
- Nerve fascicles: Visible at poles of tumor (displaced, not infiltrated)
- Ancient schwannoma: Long-standing tumors with degenerative atypia
- Cystic change: Central cystic degeneration common in large tumors
- Hyalinization: Thick-walled hyalinized vessels
- Hemorrhage: Old hemorrhage with hemosiderin deposition
Microscopic Features - Antoni A and Antoni B Pattern



Antoni A Areas (Cellular)
- Highly cellular spindle cells in fascicles
- Compact arrangement with little stroma
- Palisading nuclei forming Verocay bodies (pathognomonic)
- Nuclear palisading around anuclear eosinophilic zones
- Rows of palisaded nuclei with intervening anuclear zone
- Highly specific for schwannoma (not seen in neurofibroma)
- May be sparse or absent in some schwannomas
- Elongated spindle cells with wavy, buckled nuclei
- Pointed nuclear ends (tapered)
- Minimal cytoplasm
- No significant atypia (unless ancient schwannoma)
Antoni A areas dominate most schwannomas and contain the characteristic Verocay bodies.
Critical distinction: Ancient schwannomas show degenerative nuclear atypia (bizarre hyperchromatic nuclei) but are benign. Key features confirming benign nature:
- Low mitotic activity (less than 4 mitoses per 10 high-power fields)
- S100 diffuse positive
- No necrosis
- No infiltrative growth
Do NOT misdiagnose as malignant peripheral nerve sheath tumor (MPNST) based on atypia alone.
Beyond ancient and plexiform, two histological variants are high-yield because they are benign but are repeatedly over-called as malignant.
a highly cellular tumour composed almost entirely of Antoni A tissue with few or no Verocay bodies, with increased cellularity and sometimes brisk mitoses - so it is frequently mis-diagnosed as MPNST or a spindle-cell sarcoma. It favours deep, axial sites (retroperitoneum, pelvis, mediastinum, paravertebral region). Features confirming it is benign: diffuse strong S100 (MPNST loses S100), an intact pericellular basement membrane (collagen IV/laminin), a peripheral lymphocytic cuff and a capsule, subcapsular foamy macrophages, and the absence of true geographic necrosis and infiltrative growth. Do not over-call malignancy on cellularity or mitoses alone.
a pigmented, melanin-containing variant. The psammomatous melanotic form is associated with the Carney complex, and - unlike other schwannomas - it has genuine malignant potential (roughly 10 percent behave malignantly), so it warrants closer follow-up. (A benign microcystic/reticular variant, often in the GI tract, also exists.)
Exam point: cellular schwannoma is a benign great-mimic of sarcoma/MPNST - confirm with diffuse S100 plus an intact basement membrane; melanotic schwannoma links to the Carney complex and is the one variant with real malignant potential.
Clinical Assessment
History
- Painless mass: Most common presentation (60-70%)
- Tinel sign: Percussion over tumor elicits paresthesias (pathognomonic)
- Sensory symptoms: Paresthesias, numbness in nerve distribution (30-40%)
- Motor weakness: Uncommon unless large tumor or critical nerve
- Duration: Usually years (slow growth 1-2 mm/year)
- Vestibular schwannoma: Unilateral hearing loss, tinnitus, imbalance
- Spinal schwannoma: Radicular pain, myelopathy
- Sympathetic chain: Horner syndrome
- Vagus nerve: Hoarseness, dysphagia
Physical Examination
Examination Sequence
Visual assessment:
- Fusiform swelling along nerve course
- Normal overlying skin (unlike plexiform neurofibroma with skin changes)
- Multiple masses suggest NF2 or schwannomatosis
- Assess for cafe-au-lait spots (fewer in NF2 than NF1)
Characteristic findings:
- Firm, rubbery consistency (firmer than neurofibroma)
- Mobile perpendicular to nerve axis (PATHOGNOMONIC)
- Fixed parallel to nerve axis (tumor attached to nerve)
- Smooth, well-defined margins (encapsulated)
- Non-tender unless compressed or malignant
Percussion test:
- Tap directly over mass with reflex hammer
- Positive: Electric shock sensation radiating in nerve distribution
- Highly suggestive of nerve sheath tumor (schwannoma or neurofibroma)
- Sensitivity 60-70%, specificity 85-90%
Nerve function:
- Sensory: Light touch, pinprick, two-point discrimination in nerve distribution
- Motor: Strength testing of muscles supplied by nerve
- Reflexes: Assess deep tendon reflexes
- Baseline documentation essential before surgery
Mobile perpendicular to nerve, fixed parallel to nerve axis: This sign is pathognomonic for nerve sheath tumor (schwannoma or neurofibroma) and reflects tumor attachment to nerve with longitudinal extension. Other soft tissue tumors are mobile in all directions.
Investigations
Imaging Protocol
MRI Characteristics (Gold Standard)
Schwannoma MRI Protocol:
- T1-weighted pre-contrast
- T2-weighted with fat saturation
- T1-weighted post-gadolinium contrast
- STIR (short tau inversion recovery) sequences
Classic MRI Findings:
- Signal Characteristics
- Isointense to muscle
- Diagnostic Features
- Fusiform mass along nerve, split-fat sign (fat around tumor)
- Signal Characteristics
- Hyperintense (bright)
- Diagnostic Features
- Target sign: peripheral hyperintensity with central hypointensity (50-60%)
- Signal Characteristics
- Strong enhancement
- Diagnostic Features
- Heterogeneous enhancement (Antoni A enhances more than B)
- Signal Characteristics
- Hyperintense, fat suppressed
- Diagnostic Features
- Entering and exiting nerve (fascicular sign)
Target Sign (Pathognomonic):
- Peripheral T2 hyperintensity (Antoni B myxoid areas)
- Central T2 hypointensity (Antoni A cellular areas)
- Present in 50-60% of schwannomas (30% of neurofibromas)
- Highly specific when present
MRI is the gold standard for diagnosis and surgical planning.
Imaging Comparison: Schwannoma vs Neurofibroma
- Schwannoma
- 50-60% (peripheral bright, central dark)
- Neurofibroma
- 30% (less common)
- Schwannoma
- Present (fat rim around tumor)
- Neurofibroma
- Present (both have)
- Schwannoma
- Entering and exiting nerve visible
- Neurofibroma
- Fusiform nerve enlargement
- Schwannoma
- Strong heterogeneous enhancement
- Neurofibroma
- Homogeneous mild enhancement
- Schwannoma
- Eccentric to nerve
- Neurofibroma
- Concentric fusiform enlargement
Differential Diagnosis of a Nerve-Associated Mass
- Distinguishing Features
- Eccentric, encapsulated, Antoni A/B with Verocay bodies, diffuse S100
- Key Discriminator
- Mobile perpendicular but fixed parallel to nerve; enucleable plane
- Distinguishing Features
- Fusiform, unencapsulated, infiltrates fascicles, patchy S100, axons within tumour
- Key Discriminator
- No cleavage plane; NF1 association; concentric on imaging
- Distinguishing Features
- Large, rapidly growing, painful, new neurological deficit; loss of target sign, necrosis, heterogeneous enhancement
- Key Discriminator
- Red-flag features; arises de novo or from plexiform neurofibroma in NF1
- Distinguishing Features
- Intraneural fusiform nerve enlargement, EMA positive, S100 negative
- Key Discriminator
- EMA positive / S100 negative immunophenotype
- Distinguishing Features
- Cystic, transilluminates, fluid signal on MRI, no solid enhancement
- Key Discriminator
- Cystic non-enhancing fluid; often near joint (e.g. common peroneal at fibular neck)
- Distinguishing Features
- Fat signal on all MRI sequences; mobile in all planes (simple lipoma)
- Key Discriminator
- Follows fat on every sequence; fibrolipomatous hamartoma shows coaxial-cable nerve appearance
- Distinguishing Features
- History of nerve injury/amputation, painful Tinel, small bulbous nerve end
- Key Discriminator
- Antecedent trauma or surgery at the site
A nerve sheath mass that is greater than 5 cm, deep, rapidly enlarging, newly painful, or causing a progressive motor deficit should be treated as a malignant peripheral nerve sheath tumour until proven otherwise. Loss of a previously present target sign, intratumoral necrosis, and ill-defined margins on MRI are warning signs mandating sarcoma-unit referral and image-guided biopsy rather than enucleation.
BILATERALNF2 Diagnostic Criteria (Modified National Institutes of Health)
Hook:BILATERAL vestibular schwannomas are the hallmark of NF2 - chromosome 22 (NF-TWO)!
Management
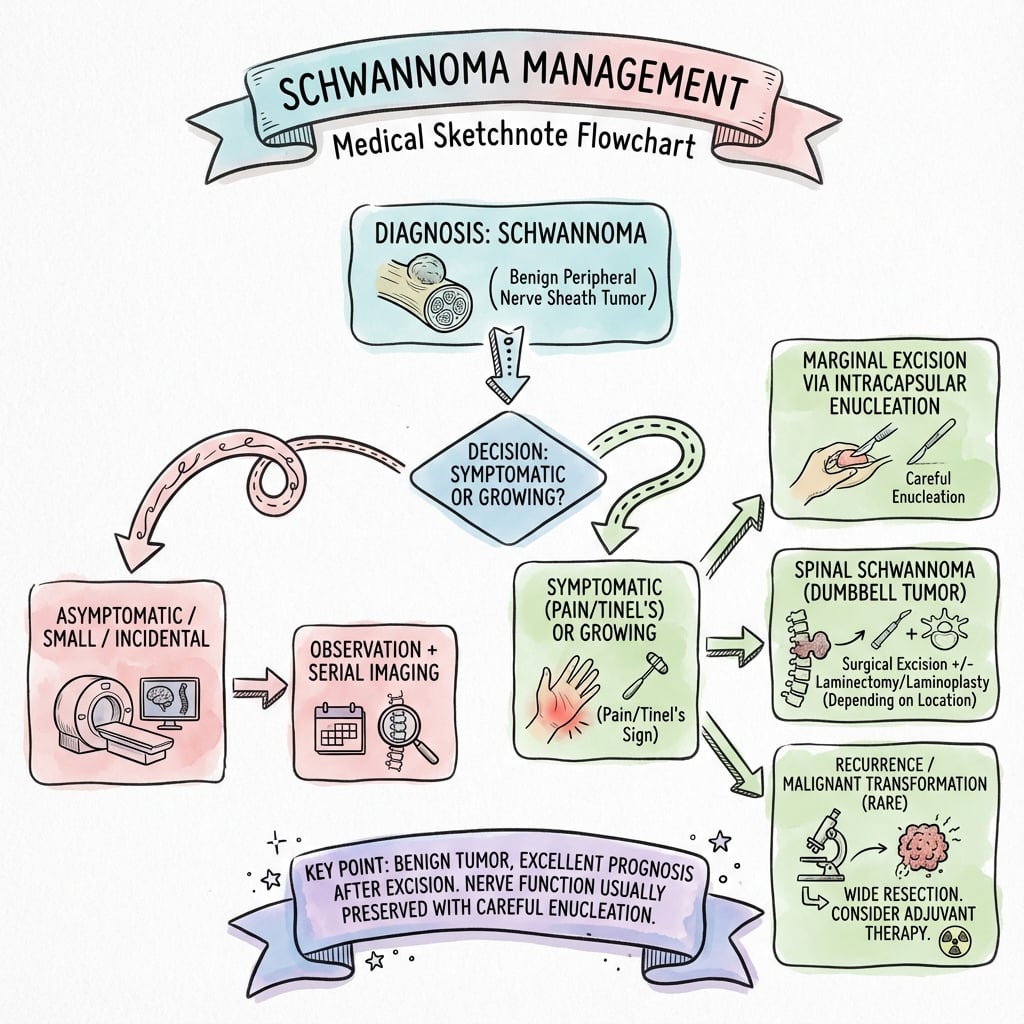
Treatment Algorithm
- Management
- Observation with annual MRI
- Rationale
- Slow growth (1-2 mm/year), low malignant potential
- Management
- Microsurgical enucleation
- Rationale
- 95% nerve preservation, less than 5% recurrence
- Management
- Observation with serial MRI vs radiosurgery
- Rationale
- Slow growth, hearing preservation with observation
- Management
- Microsurgical excision
- Rationale
- Prevent brainstem compression, decompress facial nerve
- Management
- Selective surgery with hearing preservation techniques
- Rationale
- Bilateral deafness devastating; consider cochlear implant
Surgical Technique
Microsurgical Enucleation Technique
Step-by-Step Surgical Technique
- Based on nerve location (e.g., supine for upper extremity)
- Arm/leg extended on arm board
- Tourniquet for extremity (time-limited use)
- Pad pressure points
- Longitudinal incision centered over palpable mass
- Adequate length (2-3 times tumor diameter) for exposure
- Identify and protect cutaneous nerves
- Develop subcutaneous flaps
- Identify normal nerve proximal to tumor (easier dissection)
- Identify nerve distal to tumor
- Trace nerve into tumor
- Confirm nerve continuity with nerve stimulator
- Identify adjacent vessels
- Protect vascular supply to nerve
- Ligate tumor blood supply if large feeding vessels
- Longitudinal epineurial incision over tumor
- Identify tumor capsule (glistening, distinct from nerve)
- Spread epineurium to expose tumor-nerve interface
- Nerve fascicles displaced to periphery (eccentric growth)
- Test displaced fascicles before dissection
- Identify functioning motor fascicles (stimulation causes muscle twitch)
- Sensory fascicles (no motor response)
- Document functioning fascicles
- Blunt dissection between tumor capsule and nerve fascicles
- Use microdissector, nerve hooks, small cottonoids
- Tumor shells out with gentle traction
- Preserve all functioning fascicles identified on nerve stimulator
- Enucleate entire tumor maintaining capsule integrity
- Check proximal and distal poles for residual tumor
- Small feeding vessels to tumor: bipolar cautery
- Hemostasis with bipolar (avoid nerve damage)
- Confirm all fascicles intact
- No tension on nerve
- No kinking or compression
- Test nerve stimulator again (fascicles should still function)
- Epineurium can be loosely reapproximated (optional, 7-0 nylon)
- Soft tissue closure in layers
- No drain typically needed
- Bulky dressing, splint if appropriate
Microsurgical enucleation is the gold standard for peripheral schwannomas.
Complications
Intraoperative Complications
- Incidence
- 5-10% intraoperative recognition
- Prevention
- Microsurgical technique, nerve stimulator use
- Management
- Primary repair if transected; observation if stretch injury
- Incidence
- 2-5% (higher in large vascular tumors)
- Prevention
- Bipolar cautery, tourniquet if extremity
- Management
- Hemostasis with bipolar, rarely requires transfusion
- Incidence
- 5% (residual capsule fragments)
- Prevention
- Complete visualization of tumor poles
- Management
- Observation if minimal; re-excision if symptomatic recurrence
Postoperative Complications
- Incidence
- 20-30% (neuropraxia)
- Risk Factors
- Large tumor, extensive dissection, manipulation
- Management
- Reassurance, physiotherapy; recovers 3-6 months in 90%
- Incidence
- 1-2%
- Risk Factors
- Critical nerve (facial, recurrent laryngeal), tumor adhesion to fascicles
- Management
- Occupational therapy, tendon transfers, nerve reconstruction
- Incidence
- 10-15%
- Risk Factors
- Nerve manipulation, deafferentation
- Management
- Gabapentin, pregabalin, nerve blocks; usually improves
- Incidence
- Less than 5% if complete
- Risk Factors
- Incomplete excision, plexiform variant
- Management
- Re-excision if symptomatic; observation if asymptomatic
- Incidence
- 2-3%
- Risk Factors
- Prolonged surgery, diabetes
- Management
- Antibiotics, drainage if abscess
- Incidence
- 2-3%
- Risk Factors
- Inadequate hemostasis, anticoagulation
- Management
- Observation if small; evacuation if compressive
Postoperative Care
Postoperative Timeline
- Neurovascular checks every 2-4 hours
- Document motor and sensory function
- Compare to preoperative baseline
- Watch for hematoma (rare but can compress nerve)
- Multimodal analgesia (paracetamol, NSAID, opioid PRN)
- Neuropathic pain possible (gabapentin or pregabalin)
- Ice application to reduce swelling
- Limb elevation if extremity surgery
- Gentle range of motion exercises (avoid stretching nerve)
- Splint immobilization if nerve tension concern (2 weeks)
- No heavy lifting or strenuous activity
- Keep dry for 48 hours
- Shower after 48 hours, no baths for 2 weeks
- Inspect for signs of infection
- Physiotherapy referral for motor deficits
- Desensitization for sensory changes
- Gradual return to activities
- Scar massage after suture removal
- Suture removal 10-14 days
- Histology review: Confirm schwannoma diagnosis
- Assess nerve function improvement or deficit
Surveillance:
- MRI at 6 months to confirm complete excision
- Annual clinical exam for 5 years
- Patient education: Recurrence less than 5%, watch for new mass
- Nerve function usually improves over 3-6 months (if any deficit)
Prognosis and Outcomes
Outcome by Tumor Location
- Enucleation Success
- 95%
- Nerve Preservation
- 95% preserved, 5% transient, 1% permanent deficit
- Recurrence
- Less than 5%
- Enucleation Success
- 90-95%
- Nerve Preservation
- 90-95% preserved (sciatic nerve higher risk)
- Recurrence
- 5-10%
- Enucleation Success
- 95-100%
- Nerve Preservation
- 98-100% if dorsal root (sacrifice tolerated), 85-90% if ventral root
- Recurrence
- Less than 5%
- Enucleation Success
- Complete excision 90-95%
- Nerve Preservation
- Facial nerve 70-90% preserved; hearing 10-60% preserved
- Recurrence
- 5-10% (higher for subtotal excision)
- Enucleation Success
- Subtotal excision often
- Nerve Preservation
- Variable (preserve function over complete excision)
- Recurrence
- 20-30%
Guidelines, Registries & Global Practice
Global Epidemiology
Schwannomas are encountered worldwide and are among the most common benign peripheral nerve tumours. Vestibular schwannoma incidence has risen across high-income health systems over recent decades, largely because of widespread MRI use detecting small intrameatal tumours; reported incidence ranges from roughly 1 to 2 per 100,000 person-years, with most contemporary tumours being small at diagnosis. According to PubMed, the Copenhagen national cohort cited incidence approaching 20 per million per year and showed that the majority of observed vestibular schwannomas never grow (DOI). NF2 is rare and broadly consistent across populations at approximately 1 in 33,000 births (DOI).
Side-by-Side Guideline and Society Guidance
- Scope
- Vestibular schwannoma systematic guidelines
- Core Position
- Observation, microsurgery and radiosurgery are all acceptable; serial MRI for small tumours, intervention for growth/symptoms
- Evidence Basis
- Systematic review / Level 2-3 evidence
- Scope
- NF2 and schwannomatosis diagnosis
- Core Position
- Integrate clinical features with NF2/SMARCB1/LZTR1 genetic testing; bilateral vestibular schwannomas remain diagnostic of NF2
- Evidence Basis
- Consensus guideline (Delphi)
- Scope
- Suspicious soft-tissue / nerve masses
- Core Position
- Refer indeterminate or deep/large or growing masses to a specialist sarcoma unit before biopsy or excision
- Evidence Basis
- Guideline / expert consensus
- Scope
- Soft-tissue lump red flags
- Core Position
- Urgent imaging and sarcoma-unit referral for masses greater than 5 cm, deep to fascia, rapidly growing or painful
- Evidence Basis
- Guideline
The principal global-practice safeguard is to avoid unplanned excision ("whoops" surgery) of a mass that turns out to be a malignant peripheral nerve sheath tumour. A nerve-associated mass that is large, deep, rapidly growing, painful, or shows loss of the target sign / new enhancement should be imaged with MRI and discussed at a sarcoma multidisciplinary meeting before definitive surgery, in line with NICE/BOA and EMSOS referral principles.
Practice Variation and Registry Evidence
- Peripheral schwannoma: managed by plastic, orthopaedic/hand, or neurosurgery depending on region and nerve involved
- Vestibular schwannoma: growing international shift from primary microsurgery toward observation and radiosurgery for small-to-medium tumours
- Resource-limited settings: later presentation with larger tumours; access to MRI and intraoperative neuromonitoring varies widely
- Genetic registers (e.g. UK NF2 register) underpin the epidemiological estimates and enable lifelong surveillance of affected families
- Specialist skull-base / vestibular schwannoma databases track facial-nerve, hearing and tumour-control outcomes across surgery vs radiosurgery
- NF2 / schwannomatosis multidisciplinary clinics coordinate genetic testing, hearing preservation and emerging medical therapy (e.g. bevacizumab for NF2-related vestibular schwannomas)
Referral and Medicolegal Considerations
- Primary care: GP to plastic surgery, orthopaedic/hand surgery, or neurosurgery
- Peripheral nerve schwannomas: plastic or hand surgery (upper limb), orthopaedics (lower limb)
- Vestibular schwannomas: skull-base neurosurgery or ENT (specialist centres)
- Red-flag mass: sarcoma-unit referral before biopsy if large, deep, growing or painful
- Informed consent: discuss 95% nerve preservation, 1-2% permanent deficit, less than 5% recurrence
- Documentation: preoperative nerve-function baseline essential
- Differential diagnosis: document schwannoma vs neurofibroma distinction
- NF2/schwannomatosis screening: family history, bilateral vestibular schwannomas, multiple schwannomas
- Failure to obtain baseline nerve function documentation before surgery
- Permanent nerve deficit without consent discussion of this risk
- Incomplete excision with recurrence (counsel about less than 5% recurrence vs neurofibroma 40-60%)
- Missed NF2 diagnosis (failure to ask family history, no contralateral MRI for vestibular schwannoma)
- Detailed preoperative neurological exam documented
- Informed consent including risks: transient deficit 20-30%, permanent 1-2%, recurrence less than 5%
- Intraoperative nerve stimulator use documented
- Postoperative nerve function compared to baseline
MCQ Practice Points
Q: What is the pathognomonic histological feature of schwannoma?
A: Verocay bodies - Rows of palisaded nuclei (Antoni A areas) with intervening anuclear eosinophilic zones. This feature is highly specific for schwannoma and not seen in neurofibroma. Schwannomas also show diffuse strong S100 positivity (nearly 100% of cells) compared to patchy S100 in neurofibroma (30-50%).
Q: What is the genetic basis of NF2 and what is the hallmark tumor?
A: NF2 gene on chromosome 22q12 (encodes merlin/schwannomin tumor suppressor). Bilateral vestibular schwannomas are pathognomonic for NF2 (nearly 100% of NF2 patients by age 60). Autosomal dominant inheritance, 50% penetrance by age 30. Distinguish from NF1 (neurofibromas, cafe-au-lait spots, chromosome 17).
Q: What is the target sign on MRI and what is its significance?
A: Peripheral T2 hyperintensity with central T2 hypointensity on MRI. Reflects Antoni B (hypocellular myxoid) peripherally and Antoni A (cellular) centrally. Seen in 50-60% of schwannomas vs 30% of neurofibromas. Highly specific for benign peripheral nerve sheath tumor when present.
Q: What is the success rate of nerve-sparing enucleation for schwannomas?
A: 95% nerve function preservation with microsurgical enucleation. Schwannomas are eccentric and encapsulated, displacing nerve fascicles peripherally, which creates a cleavage plane. Recurrence less than 5% with complete excision. Compare to neurofibroma which infiltrates fascicles and usually requires nerve sacrifice for complete excision.
Q: What clinical examination finding is pathognomonic for peripheral nerve sheath tumor?
A: Mobile perpendicular to nerve axis, fixed parallel to nerve axis. This sign reflects tumor attachment to nerve with longitudinal extension along nerve course. Other soft tissue tumors (lipoma, ganglion) are mobile in all directions. Tinel sign (paresthesias on percussion) is also highly suggestive but less specific.
Exam Viva Scenarios
Practise clinical reasoning and management decisions out loud
“A 45-year-old woman presents with a 3 cm painless mass in her upper arm along the radial nerve that has been slowly growing over 3 years. On examination, you find a firm mass that is mobile perpendicular to the nerve axis but fixed parallel to it. Tinel sign is positive. MRI shows a fusiform T2 hyperintense lesion with target sign and eccentric location relative to the nerve. What is your assessment and management?”
“A 32-year-old man presents with progressive right-sided hearing loss and tinnitus over 2 years. Audiometry shows right sensorineural hearing loss. MRI shows a 2.5 cm right cerebellopontine angle mass consistent with vestibular schwannoma with brainstem compression. His family history is notable for his mother having bilateral hearing loss diagnosed as vestibular schwannomas. What are your concerns and how would you manage this patient?”
“You have made a longitudinal incision and identified a 4 cm mass along the median nerve at the forearm. You suspect schwannoma based on MRI target sign. However, after epineurial incision, you find that nerve fascicles appear to run through the tumor rather than being displaced to the periphery. How would you proceed?”
Key Definition
- Benign peripheral nerve sheath tumor from Schwann cells
- Eccentric growth displaces (not infiltrates) nerve fascicles
- Well-encapsulated allows microsurgical enucleation 95% success
- 90% solitary sporadic, 10% associated NF2 (bilateral vestibular pathognomonic)
Pathognomonic Features
- Mobile perpendicular to nerve, fixed parallel (clinical exam)
- Tinel sign positive (percussion elicits paresthesias)
- Target sign on MRI: peripheral T2 bright, central dark (50-60%)
- Verocay bodies histology: palisaded nuclei with anuclear zones (Antoni A)
- S100 diffuse strong positive (nearly 100% cells)
Schwannoma vs Neurofibroma
- Schwannoma: eccentric, capsule, enucleate, S100 diffuse, NF2 association
- Neurofibroma: infiltrates, no capsule, nerve sacrifice, S100 patchy, NF1 association
- Schwannoma recurrence less than 5%; neurofibroma 40-60%
- Schwannoma preserve nerve 95%; neurofibroma usually requires sacrifice
Histology (High Yield)
- Antoni A: cellular, palisaded nuclei, Verocay bodies
- Antoni B: hypocellular, myxoid stroma, degenerative
- Ancient schwannoma: degenerative atypia but BENIGN (do not confuse with MPNST)
- S100 diffuse positive (vs patchy 30-50% in neurofibroma)
NF2 (Chromosome 22q12)
- Bilateral vestibular schwannomas pathognomonic (100% by age 60)
- Autosomal dominant, merlin/schwannomin gene loss
- Multiple peripheral schwannomas, meningiomas (45-58%), ependymomas (33%)
- Juvenile cataracts 60-81%, fewer cafe-au-lait than NF1
- Hearing preservation paramount (bilateral deafness devastating)
Surgical Technique
- Microsurgical enucleation: epineurial incision, identify capsule
- Nerve stimulator map functioning fascicles (0.5-1.0 mA)
- Blunt dissection between capsule and displaced fascicles
- 95% nerve preservation, less than 5% recurrence
- Transient deficit 20-30% (neuropraxia), permanent 1-2%
Vestibular Schwannomas
- 8% of intracranial tumors, unilateral hearing loss/tinnitus
- Observation safe for less than 1.5 cm (growth 1-2 mm/year 50%, stable 30%)
- Surgery or radiosurgery for growth/symptoms/brainstem compression
- Facial nerve preservation 70-90%, hearing 40-60% (retrosigmoid, small tumors)
Evidence Base and Key Studies
397 Peripheral Nerve Sheath Tumours: 30-Year Surgical Experience (LSUHSC Series)
- Retrospective review of 397 peripheral neural sheath tumours surgically treated 1969-1999
- 361 tumours (91%) were benign; schwannomas and neurofibromas were the dominant subtypes
- Schwannomas distributed similarly across upper and lower extremities; neurofibromas more prevalent in upper limb
- Brachial plexus was the most common single benign tumour location
- 36 malignant peripheral nerve sheath tumours (28 neurogenic sarcomas) carried high morbidity and mortality despite aggressive surgery
Pathology of Peripheral Nerve Sheath Tumours: Diagnostic Overview
- Authoritative diagnostic review defining criteria for neurofibroma, schwannoma and perineurioma
- Schwannoma: encapsulated, biphasic Antoni A (cellular, Verocay bodies) and Antoni B (myxoid) architecture, diffuse strong S100
- Neurofibroma: unencapsulated, infiltrative, entrapped axons; S100 staining only a subset of cells
- Cellular, plexiform and melanotic schwannoma variants highlighted as diagnostic pitfalls that may be over-called as malignant
- Guidance provided for separating atypical neurofibroma from MPNST and recognising borderline hybrid nerve sheath tumours
The Target Sign on MRI Differentiates Benign from Malignant Nerve Sheath Tumours
- Blinded retrospective review of 23 nerve sheath tumours (neurofibromas vs MPNST)
- Target sign (central T2 hypointensity with hyperintense rim) seen in all 12 benign neurofibromas
- Target sign present in only 1 of 11 malignant peripheral nerve sheath tumours
- Good statistical separation of benign from malignant lesions using the target sign
- Loss of a target sign in a previously typical lesion should raise concern for malignant transformation
The Natural History of Vestibular Schwannoma (Copenhagen Cohort)
- Prospective growth data on 552 observed vestibular schwannomas from a national cohort of 1818 patients
- Only 17% of purely intrameatal tumours grew during observation
- 28.9% of tumours with extrameatal extension demonstrated growth (largest-diameter change greater than 2 mm)
- Tumour growth occurred almost exclusively within the first 5 years after diagnosis
- No correlation between growth and patient sex or age, supporting primary observation of small tumours
Microsurgery vs Stereotactic Radiosurgery for Vestibular Schwannoma (Prospective Comparison)
- Prospective cohort of 82 patients with unilateral vestibular schwannoma less than 3 cm (36 resection, 46 radiosurgery)
- Normal facial movement and serviceable hearing preserved significantly more often after radiosurgery at 3 months, 1 year and last follow-up
- Tumour control was equivalent between groups (100% surgery vs 96% radiosurgery, not significant)
- Surgical resection produced significant early declines in physical-function and bodily-pain quality-of-life subscales
- Authors concluded radiosurgery should be considered first-line for most small-to-medium vestibular schwannomas
Birth Incidence and Prevalence of NF2 (UK Genetic Register)
- Population-based genetic-register study from North West England
- NF2 birth incidence estimated at approximately 1 in 33,000
- NF2 disease prevalence approximately 1 in 56,161
- 56% of NF2 cases attributable to de novo mutation (highest among the tumour-prone syndromes studied)
- Provides comparative frequencies for NF1, FAP, Gorlin and von Hippel-Lindau syndromes from a single register
Updated International Diagnostic Criteria for NF2 and Schwannomatosis
- International consensus (Delphi) recommendation revising NF2 and schwannomatosis criteria
- Bilateral vestibular schwannomas, or an identical pathogenic NF2 variant in two anatomically distinct tumours, remain diagnostic of NF2
- Molecular testing (NF2, SMARCB1, LZTR1) now central to differentiating NF2 from schwannomatosis
- Criteria explicitly incorporate mosaic forms of both conditions
- Nomenclature revised; the standalone term 'neurofibromatosis 2' was retired to improve diagnostic specificity and reduce confusion with NF1