Malignant Soft Tissue Sarcoma | t(X;18) SYT-SSX Translocation | Young Adults Near Joints
- Third most common soft tissue sarcoma in young adults (after liposarcoma and MFH)
- t(X;18) translocation creating SYT-SSX fusion is pathognomonic (present in over 95%)
- Name is misleading - does NOT arise from synovium but occurs near joints in 60-70%
- Calcification present in 30% on imaging - rare for soft tissue sarcomas
- Wide excision plus adjuvant chemotherapy and radiation is standard multimodal treatment
- “SYT-SSX1 fusion (biphasic type) has worse prognosis than SYT-SSX2 fusion (monophasic)
- “Despite name, synovial sarcoma does NOT arise from joint synovium
- “Lung is most common metastatic site (90%), bone metastases in 10-20%
- “Poorly differentiated subtype has 5-year survival under 20% versus 60% for well-differentiated
t(X;18) translocation creating SYT-SSX fusion is pathognomonic - Present in over 95% of synovial sarcomas. RT-PCR or FISH testing confirms diagnosis. SYT-SSX1 fusion has worse prognosis than SYT-SSX2.
Wide excision plus chemotherapy and radiation - Unlike many soft tissue sarcomas, synovial sarcoma is chemosensitive. Ifosfamide-based chemotherapy improves survival, especially in high-risk patients.
Does NOT arise from joint synovium - Despite the name, synovial sarcoma arises from pluripotent mesenchymal cells, not synovial lining. Occurs near joints in 60-70% but can occur anywhere.
Calcification in 30% of cases - Unusual for soft tissue sarcomas but characteristic of synovial sarcoma. Appears as stippled or curvilinear calcification on X-ray and CT.
SYNOVIALSynovial Sarcoma Key Features
Hook:SYNOVIAL reminds you of all key features - from SYT-SSX fusion to Lung metastases!
BMPSynovial Sarcoma Histological Subtypes
Hook:BMP - Biphasic is Most Poor prognosis (SYT-SSX1 worse than SSX2)!
PLANSPoor Prognostic Factors in Synovial Sarcoma
Hook:Poor PLANS lead to poor outcomes in synovial sarcoma!
Overview and Epidemiology
Synovial sarcoma is a malignant soft tissue tumor characterized by the pathognomonic t(X;18) chromosomal translocation. Despite its name, synovial sarcoma does NOT arise from joint synovium but from pluripotent mesenchymal cells. It is the third most common soft tissue sarcoma in young adults (after liposarcoma and malignant fibrous histiocytoma), accounting for 5-10% of all soft tissue sarcomas.
The name "synovial sarcoma" is historically based but anatomically incorrect. The tumor was named in the early 20th century due to its microscopic resemblance to synovial tissue and frequent occurrence near joints. However, modern molecular studies confirm it arises from primitive mesenchymal cells, NOT from joint synovium. Up to 30% occur away from joints entirely.
- Age: Peak 15-35 years (adolescents and young adults)
- Gender: Equal male and female distribution
- Pediatric: 15-20% occur in children under 15 years
- Rare in elderly: Uncommon over 50 years
- Lower extremity: 60-70% (knee, ankle, foot most common)
- Upper extremity: 15-20% (hand, wrist, forearm)
- Trunk: 10-15%
- Head/neck: 5-10% (rare but reported)
Specific Anatomical Sites
- Frequency
- 30-40%
- Typical Presentation
- Deep-seated mass near joint
- Prognostic Consideration
- Standard prognosis, limb salvage usually feasible
- Frequency
- 20-30%
- Typical Presentation
- Superficial or deep mass, often calcified
- Prognostic Consideration
- Good prognosis, smaller tumors at presentation
- Frequency
- 10-15%
- Typical Presentation
- Slow-growing mass, may mimic ganglion
- Prognostic Consideration
- Good prognosis, smaller size
- Frequency
- 10-15%
- Typical Presentation
- Large at diagnosis, deep location
- Prognostic Consideration
- Worse prognosis, difficult margins
- Frequency
- 5-10%
- Typical Presentation
- Rare, complex anatomy
- Prognostic Consideration
- Worse prognosis, margin challenges
Pathophysiology and Molecular Biology
Cellular Origin and Genetics
Synovial sarcoma arises from pluripotent mesenchymal stem cells with the capacity for epithelial differentiation. The pathognomonic t(X;18) translocation drives oncogenesis.
Pathognomonic molecular hallmark present in over 95% of synovial sarcomas:
- Translocation between chromosome X (SYT gene) and chromosome 18 (SSX1 or SSX2 gene)
- Creates SYT-SSX fusion protein
- Functions as aberrant transcription factor
- Disrupts chromatin remodeling and gene expression
Two main fusion types with prognostic significance:
- SYT-SSX1 (65%): Associated with biphasic histology, worse prognosis
- SYT-SSX2 (35%): Associated with monophasic histology, better prognosis
- Rare variants: SYT-SSX4 (under 1%), similar prognosis to SSX2
RT-PCR or FISH for t(X;18) is essential for confirming diagnosis. The presence of SYT-SSX fusion is pathognomonic for synovial sarcoma. Fusion type (SSX1 vs SSX2) has prognostic significance - SSX1 associated with worse outcomes. All suspected synovial sarcomas should undergo molecular testing.
Tumor Biology and Growth Pattern
- Description
- Slow-growing initially, then rapid progression
- Clinical Implication
- Long symptom duration (months to years) before diagnosis
- Description
- Infiltrative along fascial planes and neurovascular bundles
- Clinical Implication
- Wide margins essential, neurovascular sacrifice may be needed
- Description
- High - 40-50% metastasize within 5 years
- Clinical Implication
- Lung metastases in 90%, bone in 10-20%
- Description
- Present in 30% (rare for soft tissue sarcomas)
- Clinical Implication
- Diagnostic clue on imaging
Classification and Histology
WHO Histological Classification
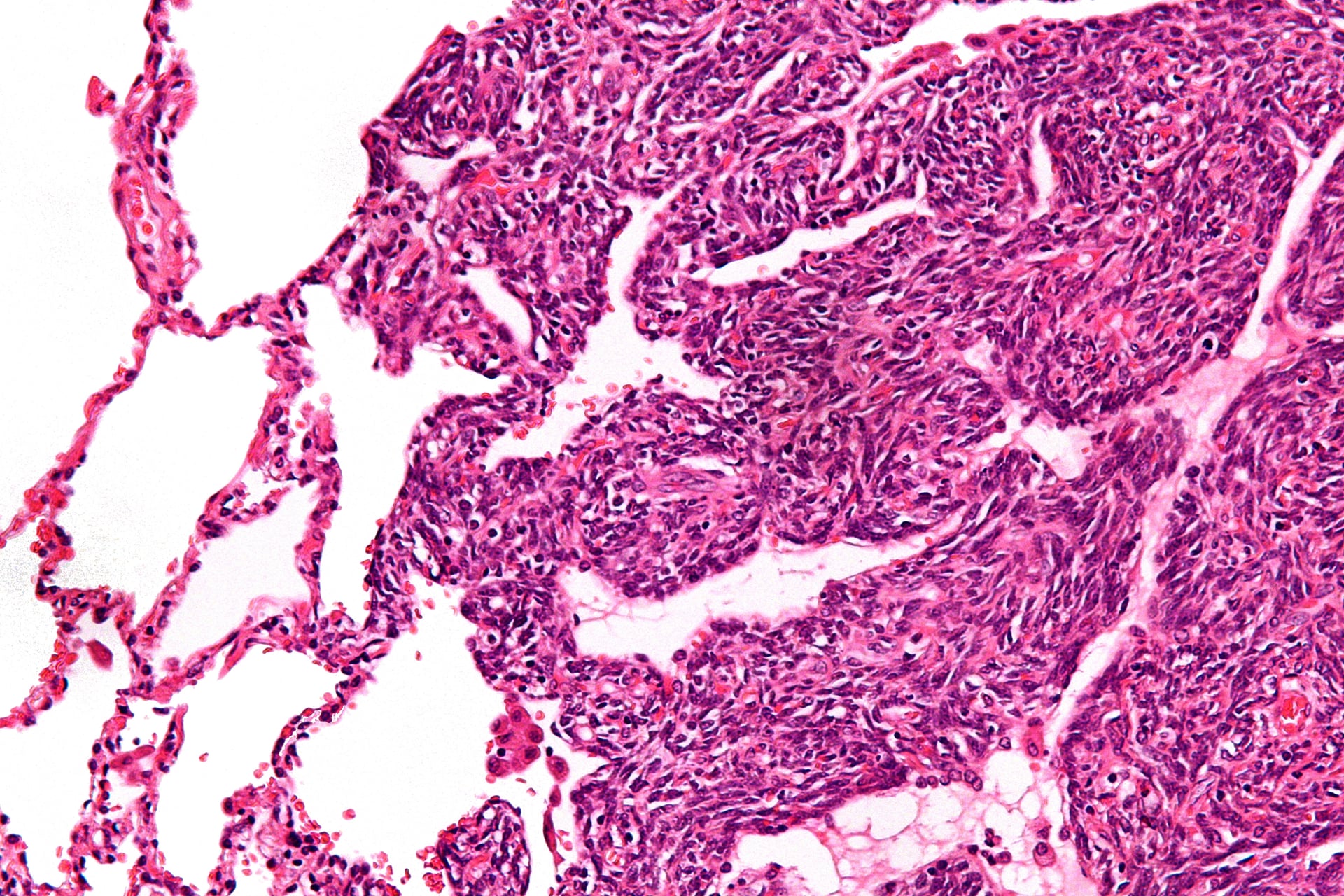
Biphasic Synovial Sarcoma (20-30%)
Classic appearance with both epithelial and spindle cell components clearly visible.
- Epithelial component: Gland-like structures, columnar or cuboidal cells
- Spindle cell component: Monotonous spindle cells in fascicles
- Two components intimately admixed
- Clear distinction between both components
- SYT-SSX1 fusion most common (over 90%)
- Worse prognosis than monophasic type
- 5-year survival: 40-50%
- Higher metastatic rate: 50-60%
The biphasic pattern is diagnostic when both components are prominent, making diagnosis easier.
Immunohistochemistry
- Positive Rate
- 70-100%
- Diagnostic Value
- Supportive but not specific
- Positive Rate
- 60-90%
- Diagnostic Value
- Supportive but not specific
- Positive Rate
- 90-95%
- Diagnostic Value
- Sensitive marker, helps distinguish from other sarcomas
- Positive Rate
- 90%
- Diagnostic Value
- Supportive but non-specific
- Positive Rate
- 60-70%
- Diagnostic Value
- May cause confusion with Ewing sarcoma
TLE1 nuclear staining is the most sensitive immunohistochemical marker, but molecular confirmation remains gold standard.
The markers above (TLE1, EMA, cytokeratin, BCL2, CD99) are supportive but not specific - TLE1 in particular is also positive in MPNST and some other sarcomas, so the topic correctly defaults to molecular confirmation. The genuine recent advance is a fusion-specific immunohistochemical antibody:
- A monoclonal antibody to the SS18-SSX fusion junction (clone E9X9V) gives strong, diffuse nuclear staining only when the fusion protein is present. In validation studies its specificity approaches 100% and sensitivity is roughly 95% for synovial sarcoma, clearly outperforming TLE1.
- A companion SSX (C-terminus) antibody can be run alongside it; concordant positivity further increases confidence.
- Practically, a positive fusion-specific stain can confirm synovial sarcoma on a small core biopsy within hours, and in many laboratories it now substitutes for FISH/RT-PCR in classic cases - molecular testing is reserved for equivocal stains or when the fusion variant (SS18-SSX1 vs SSX2) is wanted.
Exam point: when asked "how would you confirm synovial sarcoma," the modern answer is no longer just "TLE1 then molecular" - name the SS18-SSX fusion-specific antibody (E9X9V) as a highly sensitive and near-100%-specific IHC test that can confirm the diagnosis directly, with FISH/RT-PCR held in reserve for equivocal cases or fusion-variant typing.
Differential Diagnosis
Monophasic synovial sarcoma is a classic mimic of other monomorphic spindle cell tumours. The combination of clinical context, immunohistochemistry and molecular testing resolves the differential.
- Distinguishing Features
- Often arises from a nerve or in neurofibromatosis type 1; S100 focal, loss of H3K27me3
- Key Discriminator
- No SS18-SSX fusion; can be TLE1 positive so molecular test is decisive
- Distinguishing Features
- Uniform herringbone spindle cells, EMA and cytokeratin negative
- Key Discriminator
- Lacks SS18-SSX fusion and consistent TLE1 positivity
- Distinguishing Features
- Staghorn vessels, CD34 positive, STAT6 nuclear positive
- Key Discriminator
- NAB2-STAT6 fusion, not SS18-SSX
- Distinguishing Features
- Small round blue cells, diffuse membranous CD99
- Key Discriminator
- EWSR1 rearrangement; poorly differentiated synovial sarcoma can overlap, so test both
- Distinguishing Features
- Blunt-ended nuclei, eosinophilic cytoplasm, SMA/desmin positive
- Key Discriminator
- Smooth muscle markers positive, no SS18-SSX fusion
- Distinguishing Features
- Slow growth near joints; can delay diagnosis
- Key Discriminator
- Imaging features and biopsy with molecular testing exclude malignancy
A calcified soft tissue mass near a joint in a young adult is synovial sarcoma until proven otherwise. Do not be reassured by slow growth or a benign-sounding radiology report of myositis ossificans or tumoral calcinosis; biopsy with molecular testing is mandatory.
Clinical Presentation
History
- Painless mass: Most common (60-70%)
- Painful mass: 30-40% (larger tumors, neurovascular involvement)
- Slow growth: Months to years before diagnosis
- Joint symptoms: Pain, stiffness if near joint (30%)
- Average: 2-4 years before diagnosis
- Long latency: Misdiagnosed as benign (ganglion, lipoma)
- Rapid growth: Suggests high-grade or poorly differentiated
- Delay common: Slow growth leads to diagnostic delay
Average time from symptom onset to diagnosis is 2-4 years. Synovial sarcoma is frequently misdiagnosed as benign lesion (ganglion cyst, lipoma, tendon sheath tumor) due to slow growth and location near joints. Any persistent soft tissue mass in a young adult should undergo imaging and biopsy.
Physical Examination
Examination Approach
Observe:
- Size of mass (median 5cm at diagnosis)
- Location relative to joint
- Skin changes (rare except for superficial lesions)
- Venous engorgement (large tumors)
Assess:
- Consistency (firm to hard, unlike soft lipomas)
- Mobility (deep lesions often fixed to fascia)
- Tenderness (30-40% tender on palpation)
- Relation to neurovascular structures
Document:
- Range of motion of adjacent joint
- Neurovascular status distal to mass
- Muscle strength and function
- Signs of nerve compression or vascular compromise
Check:
- Lymph nodes (nodal metastases rare but possible in 5%)
- Chest examination (lung metastases uncommon at presentation)
Complete examination provides baseline for treatment planning.
Investigations and Imaging
Imaging Protocol
Plain X-rays
Initial imaging - essential for all soft tissue masses.
Key findings:
- Calcification: Present in 30% - stippled or curvilinear pattern
- Bone erosion: Adjacent bone erosion if large and juxta-osseous
- Soft tissue mass: Visible if large enough
- Periosteal reaction: Rare, indicates bone involvement
Calcification in a soft tissue mass is rare for sarcomas (occurs in under 10% overall) but is present in 30% of synovial sarcomas. This is a diagnostic clue that should raise suspicion for synovial sarcoma in young adults with soft tissue mass near a joint.
Plain radiographs are screening only - MRI is required for definitive local staging.
Biopsy
Critical rules:
- Core needle biopsy (14-16 gauge) preferred - minimally invasive
- Biopsy tract must be excisable at definitive surgery (longitudinal approach)
- Request molecular testing - RT-PCR or FISH for t(X;18) translocation
- Adequate tissue - multiple cores for histology and molecular studies
- Referral to sarcoma center before biopsy is ideal
Never perform excisional biopsy for suspected sarcoma - risks inadequate margins and tumor seeding.
Staging System
- Grade
- Low (G1)
- Size/Depth
- Superficial or deep, any size
- 5-Year Survival
- 90%
- Grade
- Low (G1)
- Size/Depth
- Deep, greater than 5cm
- 5-Year Survival
- 80%
- Grade
- Low (G1)
- Size/Depth
- Deep, greater than 10cm OR High (G2-3) under 5cm
- 5-Year Survival
- 70%
- Grade
- High (G2-3)
- Size/Depth
- Superficial/deep, 5-10cm
- 5-Year Survival
- 60%
- Grade
- High (G2-3)
- Size/Depth
- Deep, greater than 10cm
- 5-Year Survival
- 40-50%
- Grade
- Any
- Size/Depth
- Metastatic disease (M1)
- 5-Year Survival
- 10-20%
Most synovial sarcomas are intermediate to high grade (G2-G3) by virtue of their biology.
Management
Core Principles
Synovial sarcoma requires multimodal treatment: Wide surgical resection PLUS adjuvant chemotherapy PLUS radiation therapy. Unlike many soft tissue sarcomas, synovial sarcoma is chemosensitive. Ifosfamide-based chemotherapy improves survival, particularly for high-risk patients (larger than 5cm, high grade, deep location).
Treatment Fundamentals:
- Surgery: Wide excision with 2cm margins is the goal
- Chemotherapy: Ifosfamide-based regimens (chemosensitive tumor)
- Radiation: 50-66 Gy pre- or postoperative
These principles apply to all synovial sarcoma cases.

The topic states radiotherapy can be given "pre- or postoperative" and separately lists wound complications and late radiation fibrosis among the morbidities - the examinable link between them is the timing tradeoff defined by the O'Sullivan NCIC SR2 randomised trial (extremity soft tissue sarcoma).
Preoperative radiotherapy (lower dose, about 50 Gy; smaller field treating only the tumour plus margin):
- Pro: smaller radiation volume and lower total dose mean less late fibrosis, oedema and joint stiffness, and an undisturbed, better-oxygenated tumour bed; the tumour may shrink to aid resection.
- Con: roughly double the rate of major acute wound-healing complications (around 35 percent versus 17 percent in SR2), because surgery is through irradiated tissue.
Postoperative radiotherapy (higher dose, about 60-66 Gy; larger field covering the whole surgical bed and drain sites):
- Pro: surgery through non-irradiated tissue, so fewer acute wound complications; full pathology available before planning.
- Con: larger field and higher dose drive more late effects (fibrosis, oedema, stiffness, fracture) that worsen long-term limb function.
Local control is equivalent between the two; the choice is a tradeoff of early wound morbidity (worse with preop) against late functional morbidity (worse with postop), individualised at the sarcoma MDT - preop is often favoured for large/deep tumours near critical structures where field size and late function matter most.
Exam point: preop RT = lower dose/smaller field, more acute wound problems but less late fibrosis; postop RT = higher dose/larger field, fewer wound problems but more late fibrosis/stiffness; local control is the same (O'Sullivan NCIC SR2).
Surgical Technique
Preoperative Planning
Preparation Steps
- Review MRI with radiology and surgical team
- Identify tumor extent, neurovascular involvement
- Plan resection margins (2cm goal for synovial sarcoma)
- Assess need for vascular or nerve reconstruction
- Mark biopsy tract for en bloc excision
- Plan incision to incorporate biopsy site
- Avoid transverse incisions (limit extensile approaches)
- Consider drain sites as contaminated
- Anticipate defect size and depth
- Arrange soft tissue coverage (flap, graft)
- Plan tendon transfers if muscle resection needed
- Vascular surgery backup if vessel reconstruction needed
Preparation ensures optimal surgical outcome.
Complications
Treatment-Related Complications
- Incidence
- 15-30% at 5 years
- Risk Factors
- Positive margins, large size, high grade
- Management
- Re-resection if feasible, amputation for failed limb salvage
- Incidence
- 40-50% at 5 years
- Risk Factors
- High grade, larger than 5cm, poorly differentiated
- Management
- Systemic chemotherapy, metastasectomy if oligometastatic
- Incidence
- 15-30% (higher with preop RT)
- Risk Factors
- Preoperative radiation, poor soft tissue coverage
- Management
- Wound care, VAC therapy, flap coverage if needed
- Incidence
- 10-20%
- Risk Factors
- Neurovascular proximity, intentional sacrifice
- Management
- Physiotherapy, orthotics, nerve reconstruction if feasible
- Incidence
- 20-40% at 10 years
- Risk Factors
- High radiation dose, large field
- Management
- Physiotherapy, stretching, manage expectations
- Incidence
- 50-80% (varies by regimen)
- Risk Factors
- High-dose ifosfamide, doxorubicin cumulative dose
- Management
- Supportive care, growth factors, dose modification
Postoperative Care and Surveillance
Recovery Protocol
Postoperative Management
Inpatient care:
- Wound monitoring (flap viability, hematoma, infection)
- DVT prophylaxis (LMWH)
- Pain management (multimodal analgesia)
- Drain management (remove when under 30ml per 24 hours)
Clinic review:
- Wound healing assessment
- Suture removal at 2-3 weeks
- Final pathology review (margins, grade, molecular)
- Plan adjuvant therapy (chemotherapy and/or radiation)
Chemotherapy and radiation:
- Chemotherapy cycles (4-6 cycles over 3-4 months)
- Radiotherapy (5-7 weeks, 50-66 Gy)
- Monitor for treatment toxicity
- Maintain physiotherapy during treatment
Follow-up schedule:
- Years 1-2: Every 3 months
- Years 3-5: Every 6 months
- Beyond 5 years: Annually
Imaging and surveillance detailed below.
Surveillance Protocol
Every 3 months:
- Clinical examination
- Chest CT (detect lung metastases early)
- MRI primary site every 6 months
- 70% of recurrences occur in first 3 years
Every 6 months:
- Clinical examination
- Chest CT every 6 months
- MRI primary site annually
- Late recurrences possible
Annually:
- Clinical examination
- Chest imaging (X-ray or CT)
- Local imaging if symptoms
- Late metastases can occur 10+ years
High metastatic rate requires vigilant imaging:
- Lung CT: High-resolution for nodule detection
- PET-CT: If recurrence suspected, assess resectability
- Bone scan: If bone pain (10-20% bone metastases)
Prognosis and Outcomes
Prognostic Factors
- Favorable
- Monophasic, SYT-SSX2
- Unfavorable
- Poorly differentiated, SYT-SSX1
- Favorable
- Under 5cm
- Unfavorable
- Greater than 5cm
- Favorable
- Superficial
- Unfavorable
- Deep (subfascial)
- Favorable
- Under 25 years
- Unfavorable
- Over 25 years
- Favorable
- Negative (greater than 2mm)
- Unfavorable
- Positive
- Favorable
- SYT-SSX2
- Unfavorable
- SYT-SSX1
- Favorable
- Greater than 90% necrosis
- Unfavorable
- Poor response
Survival by Subtype and Stage
- 5-Year Survival
- 50-60%
- 10-Year Survival
- 35-45%
- 5-Year Survival
- 70-80%
- 10-Year Survival
- 55-65%
- 5-Year Survival
- 50-60%
- 10-Year Survival
- 35-45%
- 5-Year Survival
- Under 20%
- 10-Year Survival
- Under 10%
- 5-Year Survival
- 10-20%
- 10-Year Survival
- Under 10%
SYT-SSX fusion type has prognostic significance: SYT-SSX1 fusion (associated with biphasic histology) has worse prognosis (5-year survival 40-50%) compared to SYT-SSX2 fusion (associated with monophasic histology, 5-year survival 60-70%). This is independent of other factors.
Guidelines, Registries & Global Practice
Global Epidemiology
- Incidence: Roughly 1 to 3 per million population per year worldwide
- Share of sarcomas: 5 to 10% of all soft tissue sarcomas
- Age: Disproportionate impact in adolescents and young adults (peak 15 to 35 years)
- Paediatric: One of the more common non-rhabdomyosarcoma soft tissue sarcomas in children
- Nodal spread is rare: SEER analysis found regional lymph-node metastasis in only 3.2% of synovial sarcomas
- Survival: 5-year overall survival roughly 50 to 70% for localized disease, far lower once metastatic
- Metastatic pattern: Lung dominant (around 90% of metastases)
Side-by-Side Guidance from Major Societies
- Diagnosis
- Refer to sarcoma centre before biopsy; molecular confirmation of SS18-SSX
- Local Treatment
- Wide excision plus radiotherapy for deep/larger tumours
- Systemic Therapy
- Anthracycline plus ifosfamide considered for high-risk and chemosensitive subtypes
- Diagnosis
- Image-guided core biopsy at a sarcoma centre; multidisciplinary review
- Local Treatment
- Limb-sparing wide excision with radiotherapy; re-excision for positive margins
- Systemic Therapy
- Neoadjuvant/adjuvant chemotherapy individualized; pazopanib for relapsed disease
- Diagnosis
- Mandatory specialist sarcoma MDT before definitive treatment
- Local Treatment
- Centralized surgery and radiotherapy at designated centres
- Systemic Therapy
- Chemotherapy reserved for selected high-risk or advanced cases
- Diagnosis
- Molecular confirmation; risk-stratified protocols
- Local Treatment
- Conservative surgery favoured to preserve function in young patients
- Systemic Therapy
- Risk-adapted ifosfamide-based chemotherapy in intermediate/high-risk groups
Every major society agrees on three points regardless of region: any suspicious deep or enlarging soft tissue mass should be referred to a specialist sarcoma centre before biopsy, diagnosis should be molecularly confirmed (SS18-SSX fusion), and definitive management should be planned by a multidisciplinary sarcoma team. Disagreement centres mainly on the threshold for adjuvant chemotherapy.
High- vs Limited-Resource Practice Variation
- Routine FISH/RT-PCR or NGS for SS18-SSX confirmation
- Centralized sarcoma MDTs and limb-salvage surgery with reconstruction
- Access to MRI staging, PET-CT and pazopanib
- HLA-A*02/MAGE-A4 testing and TCR T-cell therapy (afami-cel) at select cellular-therapy centres
- Diagnosis may rely on morphology plus TLE1 immunohistochemistry where molecular testing is unavailable
- Later presentation with larger tumours; higher amputation rates
- Restricted radiotherapy and chemotherapy access
- Telepathology and regional referral networks help bridge expertise gaps
Applicable in any health system:
- Document the investigation pathway for any persistent soft tissue mass (mitigates the common delayed-diagnosis pitfall)
- Confirm pre-biopsy staging and that biopsy was planned with the definitive surgeon
- Record that molecular confirmation was requested
- Evidence of sarcoma MDT discussion before definitive treatment
- Informed consent covering multimodal therapy, recurrence and metastatic risk, and possible amputation
- A written long-term surveillance plan, given late recurrences beyond 5 to 10 years
Controversies and Areas of Uncertainty
The single most debated issue. The Italian trial showed a survival benefit in high-risk extremity sarcoma, but pooled meta-analyses and longer follow-up are less consistent, and the EORTC 62931 trial of doxorubicin plus ifosfamide showed no overall-survival benefit. Most centres reserve chemotherapy for young patients with large, deep, high-grade tumours, decided case by case at MDT.
SYT-SSX1 versus SSX2 correlates with histology and survival in retrospective series, yet on multivariate analysis tumour size and stage often dominate. Fusion type is best regarded as one input among several rather than a standalone treatment driver.
Resection of pulmonary metastases is widely practised for oligometastatic, resectable disease and is associated with longer survival in selected patients, but no randomized trial confirms a survival benefit. Patient selection (resectability, disease-free interval, fitness) is decisive.
Engineered TCR T-cell therapy (afami-cel) is a genuine advance but is limited to HLA-A*02-positive, MAGE-A4-expressing tumours and to specialist centres, raising equity-of-access questions globally.
MCQ Practice Points
Q: What chromosomal translocation is pathognomonic for synovial sarcoma? A: t(X;18) translocation creating SYT-SSX fusion - Present in over 95% of synovial sarcomas. The translocation fuses the SYT gene on chromosome X with either SSX1 or SSX2 gene on chromosome 18. SYT-SSX1 fusion has worse prognosis than SYT-SSX2. RT-PCR or FISH confirms diagnosis.
Q: What is the peak age and most common location for synovial sarcoma? A: Peak age 15-35 years (young adults), most common near large joints (knee, ankle, foot) in 60-70%. Despite the name, synovial sarcoma does NOT arise from joint synovium but from mesenchymal cells. It is the third most common soft tissue sarcoma in young adults.
Q: What imaging finding is characteristic of synovial sarcoma but rare for other soft tissue sarcomas? A: Calcification - present in 30% of synovial sarcomas but rare (under 10%) in most other soft tissue sarcomas. Appears as stippled or curvilinear calcification on X-ray and CT. MRI triple sign (three signal intensities on T2) is also characteristic.
Q: How does treatment of synovial sarcoma differ from most other soft tissue sarcomas? A: Synovial sarcoma is chemosensitive and requires multimodal treatment: surgery PLUS chemotherapy PLUS radiation. Ifosfamide-based chemotherapy improves survival in high-risk patients. Response rate is 30-50%, better than most soft tissue sarcomas. Wide excision alone is insufficient.
Q: What is the 5-year survival for synovial sarcoma and what is the most common site of metastasis? A: Overall 5-year survival is 50-60%. Lungs are the most common metastatic site (90% of metastases). Metastatic rate is 40-50% at 5 years. Poor prognostic factors include: size greater than 5cm, poorly differentiated histology, SYT-SSX1 fusion, positive margins.
Exam Viva Scenarios
Practise clinical reasoning and management decisions out loud
“A 22-year-old woman presents with a 4cm painless mass on the medial aspect of her ankle, present for 18 months and slowly enlarging. MRI shows a multiloculated soft tissue mass with triple sign on T2 imaging and small areas of calcification on X-ray. What is your differential diagnosis and management approach?”
“A 28-year-old man has biopsy-proven synovial sarcoma of the thigh (8cm, deep to fascia, SYT-SSX1 fusion, biphasic histology). CT chest shows no metastases. The tumor is adjacent to the femoral neurovascular bundle. How would you manage this case?”
“A 30-year-old woman treated 2 years ago for synovial sarcoma of the foot (wide excision, chemotherapy, radiation) now presents with 3 lung nodules on surveillance CT (largest 2cm, all in right lung). PET-CT shows FDG-avid nodules, no other disease. How do you proceed?”
Key Epidemiology
- Third most common soft tissue sarcoma in young adults (5-10% of all soft tissue sarcomas)
- Peak age 15-35 years, equal male and female distribution
- 60-70% near large joints (knee, ankle, foot), but does NOT arise from synovium
- Misleading name - arises from mesenchymal cells not synovial tissue
Molecular Biology (Pathognomonic)
- t(X;18) translocation creating SYT-SSX fusion present in over 95%
- SYT-SSX1 fusion (65%): Biphasic histology, worse prognosis (5-year survival 40-50%)
- SYT-SSX2 fusion (35%): Monophasic histology, better prognosis (5-year survival 60-70%)
- RT-PCR or FISH confirms diagnosis - molecular testing essential
Histological Classification
- Biphasic (20-30%): Epithelial AND spindle components visible
- Monophasic (60-70%): Spindle cells only, epithelial component hidden
- Poorly differentiated (10-15%): High-grade, under 20% 5-year survival
- Immunohistochemistry: TLE1 (90-95% sensitive), EMA, cytokeratin positive
Clinical Presentation and Imaging
- Painless slow-growing mass (60-70%), average 2-4 years symptom duration
- Calcification in 30% on imaging - rare for soft tissue sarcomas (diagnostic clue)
- MRI triple sign: Three signal intensities on T2 (characteristic)
- CT chest essential for staging - lungs are site of 90% of metastases
Treatment Principles (Multimodal)
- Wide excision with 2cm margins (negative margins critical)
- Chemotherapy: Ifosfamide-based regimens (synovial sarcoma is chemosensitive, response rate 30-50%)
- Radiotherapy: 50-66 Gy adjuvant for high-risk or close margins
- Multimodal treatment (surgery plus chemo plus radiation) is standard unlike many sarcomas
- Neoadjuvant chemotherapy for large (greater than 5cm) or high-risk tumors
Poor Prognostic Factors (PLANS Mnemonic)
- Poorly differentiated histology (under 20% 5-year survival)
- Large size (greater than 5cm)
- Age over 25 years
- Neurovascular invasion
- SYT-SSX1 fusion type (worse than SSX2)
Prognosis and Surveillance
- Overall 5-year survival: 50-60%, 10-year survival: 35-45%
- Metastatic rate: 40-50% at 5 years (lungs 90%, bone 10-20%)
- Local recurrence: 15-30% (negative margins essential)
- Surveillance: Every 3 months years 1-2, every 6 months years 3-5, annually after 5 years
- Chest CT critical - 70% of metastases occur within first 3 years
Evidence Base and Key Studies
Discovery of the t(X;18) SYT-SSX Fusion
- Cloned the genes spanning the recurrent t(X;18)(p11.2;q11.2) breakpoint in synovial sarcoma
- Identified the novel SYT gene on chromosome 18 fused to the SSX gene on the X chromosome
- Demonstrated genomic SYT rearrangement in 10 of 13 synovial sarcomas tested
- Showed rearrangement generates an in-frame SYT-SSX fusion transcript acting as an aberrant transcriptional regulator
SYT-SSX Fusion Type Determines Morphology and Prognosis
- RT-PCR analysis of 45 synovial sarcomas: SYT-SSX1 in 64% and SYT-SSX2 in 36%
- All 12 biphasic tumours carried SYT-SSX1; all SYT-SSX2 tumours were monophasic (P=0.003)
- In localized disease, SYT-SSX2 had significantly better metastasis-free survival than SYT-SSX1 (relative risk 3.0, P=0.03 multivariate)
- Histologic subtype alone was not independently prognostic once fusion type was considered
Fusion Type in a Multi-Institutional Cohort of 243 Patients
- 243 patients across multiple institutions: SYT-SSX1 in 61% and SYT-SSX2 in 37%
- Median and 5-year overall survival: SYT-SSX1 6.1 years and 53% versus SYT-SSX2 13.7 years and 73%
- Fusion type was the single most significant prognostic factor on multivariate analysis in localized disease
- Strong association of fusion type with morphology (almost all biphasic tumours were SYT-SSX1)
Prognostic Factors in Primary Localized Extremity Synovial Sarcoma
- 112 patients with primary localized extremity synovial sarcoma, median follow-up 72 months
- 5-year local-recurrence, distant-recurrence and mortality rates were 12%, 39% and 25% respectively
- Tumour size 5cm or greater was an independent adverse predictor of distant recurrence and mortality (RR ~2.3-2.7)
- Bone or neurovascular invasion was independently associated with distant recurrence and death
Italian Adjuvant Chemotherapy Trial (High-Grade Extremity STS)
- Randomized trial of 104 patients with high-grade (G3-4) spindle-cell extremity/girdle sarcomas 5cm or larger
- Five cycles of epirubicin plus ifosfamide versus observation after local therapy
- Median overall survival 75 months with chemotherapy versus 46 months for controls (P=0.03)
- Absolute overall-survival benefit 13% at 2 years rising to 19% at 4 years; no toxic deaths
TLE1 as a Diagnostic Immunohistochemical Marker
- Prospective evaluation of TLE1 in cases where synovial sarcoma was a real diagnostic consideration
- Diffuse nuclear TLE1 staining in 35 of 35 molecularly confirmed synovial sarcomas
- Rare to absent in 73 other soft tissue tumours (only isolated MPNST, fibrosarcoma and pleomorphic sarcoma positive)
- Positive predictive value 92% and negative predictive value 100% in this setting; outperformed BCL2, EMA and cytokeratin
PALETTE: Pazopanib for Metastatic Soft Tissue Sarcoma
- Phase 3 randomized, double-blind, placebo-controlled trial in 369 patients with pretreated metastatic non-adipocytic STS
- Synovial sarcoma was a major enrolled subtype (liposarcoma excluded)
- Median progression-free survival 4.6 months with pazopanib versus 1.6 months with placebo (HR 0.31, P less than 0.0001)
- Overall survival 12.5 versus 10.7 months (HR 0.86, not significant)
SPEARHEAD-1: Afami-cel TCR T-Cell Therapy
- International open-label phase 2 trial of afamitresgene autoleucel (MAGE-A4-directed TCR T cells) in HLA-A*02 patients
- 52 heavily pretreated patients (44 synovial sarcoma, 8 myxoid/round cell liposarcoma)
- Overall response rate 39% in synovial sarcoma; responses were durable
- Cytokine release syndrome in 71% (mostly low grade) and grade 3 or worse cytopenias common; no treatment-related deaths