Cleidocranial Dysplasia | RUNX2 (CBFA1) Deficiency | Defective Intramembranous Ossification
- RUNX2 (CBFA1) deficiency - haploinsufficiency of the master osteoblast transcription factor impairs intramembranous AND endochondral ossification
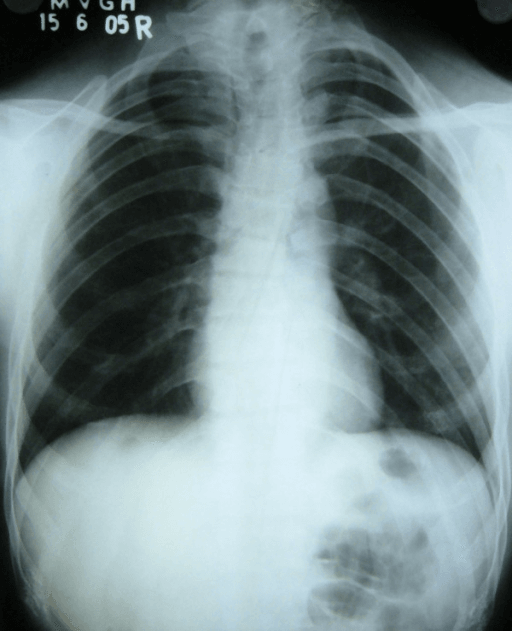
- Absent/hypoplastic clavicles - hallmark allowing abnormal approximation of the shoulders in front of the chest
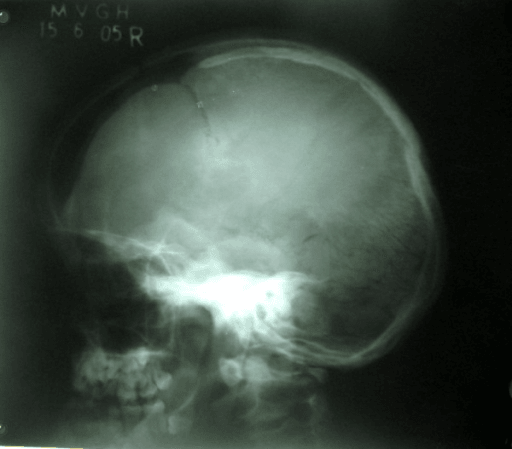
- Delayed cranial ossification - persistent open fontanelles and sutures with multiple Wormian bones
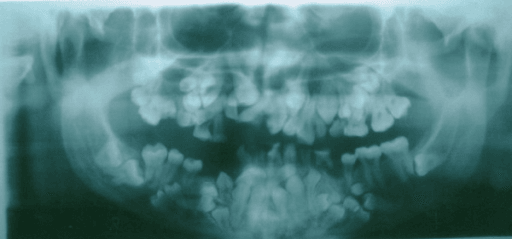
- Dental anomalies - retained deciduous teeth, failure of permanent eruption, and supernumerary teeth needing lifelong dental care
- Normal intelligence and life expectancy - this is a skeletal/dental disorder, not a neurodevelopmental one
- “Inheritance is autosomal dominant with variable expressivity; approximately 40% are de novo (no family history)
- “Clavicular involvement is often partial - the most common pattern is a defect at the junction of the lateral and middle thirds
- “Coxa vara and femoral neck deformity are the orthopaedic surgeon's main concern and may need a valgus osteotomy
- “Distinguish from pycnodysostosis - that has dense sclerotic bones and acroosteolysis; cleidocranial dysostosis has normal/reduced density
RUNX2 (CBFA1) on chromosome 6p21 is the master transcription factor that drives osteoblast differentiation and bone formation. Loss of one functional copy (haploinsufficiency) is enough to cause disease, which is why inheritance is autosomal dominant. It impairs intramembranous ossification most (clavicle, skull) but also affects endochondral bone.
Absent or hypoplastic clavicles are the clinical hallmark. The classic bedside sign is the ability to approximate the shoulders in front of the chest. Clavicular aplasia is uncommon - partial defects, typically at the junction of the middle and lateral thirds, are more frequent.
Delayed ossification of the skull leaves the fontanelles and sutures open for years, with multiple Wormian bones (small intrasutural ossicles) and frontal/parietal bossing. Brachycephaly and midface hypoplasia are typical. Intelligence is normal.
Do NOT confuse with pycnodysostosis. Cleidocranial dysostosis bones are normal or slightly reduced in density - there is no osteosclerosis and no acroosteolysis. The shared "open fontanelle" feature is a classic exam trap; the bone density on the film tells you which condition it is.
- Key Features
- Absent/hypoplastic clavicles, narrow thorax
- Management
- Reassurance; usually no clavicle surgery needed
- Exam Pearl
- Ability to approximate shoulders is the classic sign
- Key Features
- Delayed suture closure, Wormian bones, bossing
- Management
- Helmet/protective advice, monitor, genetics referral
- Exam Pearl
- Check bone density - normal here, dense in pycnodysostosis
- Key Features
- Coxa vara, femoral neck varus, possible scoliosis
- Management
- Valgus osteotomy for symptomatic/progressive coxa vara
- Exam Pearl
- Coxa vara is the main orthopaedic problem to treat
- Key Features
- Retained deciduous, supernumerary and unerupted teeth
- Management
- Lifelong orthodontic-surgical programme
- Exam Pearl
- Dental burden often dominates quality of life
CLAVICLECardinal Features of Cleidocranial Dysostosis
Hook:The CLAVICLE is the headline finding - spell it out and you have recalled the whole syndrome.
OPENDistinguishing from Pycnodysostosis
Hook:Both have OPEN fontanelles - but cleidocranial dysostosis keeps things OPEN and normal-density, while pycnodysostosis is dense and erodes the fingertips.
HIPSOrthopaedic and Dental Concerns
Hook:Watch the HIPS (and the mouth) - these are where active treatment is actually needed.
Overview and Epidemiology
Cleidocranial dysostosis (also called cleidocranial dysplasia) is a generalised skeletal dysplasia that preferentially affects bones formed by intramembranous ossification, especially the clavicle and the skull. It was historically named for the two most obvious features - "cleido" (clavicle) and "cranial" (skull). The modern term cleidocranial dysplasia is preferred because the disorder affects the whole skeleton, not just two bones. According to PubMed, it is caused by heterozygous loss-of-function variants in the RUNX2 (CBFA1) gene (DOI).
- Incidence: Approximately 1 per 1 million live births
- Gender: Affects males and females equally
- Inheritance: Autosomal dominant, full penetrance, highly variable expressivity
- De novo rate: Around 40% have no affected parent (new mutation)
- Detection: May be diagnosed in adulthood as an incidental finding
- Onset: Present at birth; often recognised in childhood
- Intelligence: Normal cognitive development
- Life expectancy: Normal
- Growth: Mild short stature is common
- Main burden: Dental disease and selected orthopaedic deformities
Why the Name Changed
The traditional name "cleidocranial dysostosis" highlights the clavicle and skull, but RUNX2 haploinsufficiency disturbs ossification throughout the skeleton - pelvis, spine, hands and long bones are all involved. For this reason most contemporary literature uses cleidocranial dysplasia. For exam purposes the two terms are interchangeable, and examiners may use either.
Spectrum of Severity
Expressivity is wide even within one family. Some individuals have the full picture of absent clavicles, delayed cranial ossification and dozens of unerupted teeth, while others have only subtle dental anomalies or mild clavicular hypoplasia. According to PubMed, missense variants within the functionally critical Runt homology domain (RHD) tend to produce more severe skeletal phenotypes than other variant types (DOI).
Pathophysiology and Genetics
RUNX2 - the Master Osteoblast Gene
According to PubMed, cleidocranial dysostosis is caused by heterozygous loss-of-function variants in RUNX2 (also called CBFA1) on chromosome 6p21, the master transcription factor controlling osteoblast differentiation and skeletal morphogenesis in vertebrates (DOI). A single defective copy is sufficient to cause disease because the skeleton is sensitive to RUNX2 dosage (haploinsufficiency). RUNX2 drives commitment of mesenchymal precursors to the osteoblast lineage, so its deficiency impairs both intramembranous and endochondral bone formation - although intramembranous bones (clavicle, skull vault) are the most visibly affected.
- Gene: RUNX2 / CBFA1 on chromosome 6p21
- Protein: Runt-related transcription factor 2
- Key domain: Runt homology domain (RHD) binds DNA and the CBFB cofactor
- Mechanism: Haploinsufficiency (loss of one functional allele)
- Variant types: Missense, nonsense, frameshift, splice-site, deletions, translocations
- Osteoblasts: Reduced differentiation and matrix production
- Intramembranous bone: Clavicle and skull vault most affected
- Endochondral bone: Delayed and disordered ossification centres
- Teeth: Defective root and eruption regulation (RUNX2 in dental follicle)
- Result: Generalised but variable skeletal under-ossification
Genotype-Phenotype Correlations
A clean genotype-phenotype correlation has historically been hard to establish, but large data sets are now refining it. According to PubMed, a 2024 systematic review of 569 variants in 453 patients found that variants cluster predominantly in the Runt homology domain (RHD), and that missense RHD variants are significantly associated with more severe features - supernumerary teeth, macrocephaly, short ribs, hypoplastic iliac wings and limited shoulder abduction - than nonsense or other in-frame variants (DOI). In-frame insertions/deletions tend to produce fewer features.
Inheritance and Counselling
Cleidocranial dysostosis is autosomal dominant with essentially full penetrance but highly variable expressivity:
- Each child of an affected person has a 50% chance of inheriting the variant.
- Approximately 40% of cases are de novo (new mutation, no family history).
- Because expressivity varies, a parent may be mildly (even subclinically) affected - examine and image the parents.
- Molecular confirmation of the family's RUNX2 variant enables predictive and prenatal testing.
Clinical Features
Cardinal Features
The classic clinical picture combines clavicular, cranial, dental and skeletal features:
- Clavicular hypoplasia or aplasia (ability to approximate the shoulders)
- Delayed closure of fontanelles and sutures with frontal/parietal bossing
- Midface hypoplasia with relative mandibular prognathism
- Dental anomalies (retained deciduous, unerupted permanent and supernumerary teeth)
- Skeletal anomalies (coxa vara, scoliosis, brachydactyly, delayed pubic ossification)
- Patent anterior fontanelle: Persists well beyond infancy
- Brachycephaly: With frontal and parietal bossing
- Wormian bones: Multiple intrasutural ossicles
- Midface hypoplasia: Flat profile, depressed nasal bridge
- High-arched palate: Sometimes with submucous/overt cleft
- Relative mandibular prognathism: Due to underdeveloped maxilla
- Clavicles: Absent or hypoplastic, often at lateral/middle third junction
- Narrow thorax: Bell-shaped, sloping shoulders
- Coxa vara: Femoral neck varus, the key orthopaedic deformity
- Spine: Scoliosis, kyphosis, spondylolysis in some patients
- Hands: Brachydactyly, tapering digits, pseudoepiphyses
- Pelvis: Delayed ossification, wide symphysis pubis
Physical Examination
- Narrow, drooping ("hangdog") shoulders that can be brought forward to touch in the midline
- Hypermobility of the shoulder girdle from clavicular deficiency
- Bell-shaped, narrow thorax
- Large head with frontal and parietal bossing
- Palpable open fontanelle/sutures (may persist into adult life)
- Flat midface, depressed nasal bridge, relative prognathism
- Retained deciduous teeth and failure of permanent eruption
- Multiple supernumerary teeth (often discovered on imaging)
- High-arched, narrow palate
- Mild short stature
- Possible coxa vara with waddling gait
- Brachydactyly and ligamentous laxity
- Scoliosis or kyphosis in a minority
Multidisciplinary Burden
According to PubMed, a longitudinal craniofacial-unit series of 14 patients found that essentially all had frontal bossing, a patent anterior fontanelle, multiple Wormian bones, midface hypoplasia, abnormal dentition, clavicular hypoplasia or aplasia and normal intellect; obstructive sleep apnoea, recurrent ear infections and speech/hearing issues were common, underscoring the need for coordinated multidisciplinary care (DOI).
Investigations and Radiographic Features
Diagnostic Imaging
The diagnosis is usually made on clinical and radiographic grounds. The triad to look for is: (1) absent or hypoplastic clavicles on the chest film; (2) delayed cranial ossification with persistent open fontanelles/sutures and multiple Wormian bones; and (3) dental anomalies with retained deciduous teeth, multiple unerupted permanent teeth and supernumerary teeth on the orthopantomogram. Bone density is normal - if the bones are dense, reconsider pycnodysostosis or osteopetrosis. Molecular confirmation of a RUNX2 variant secures the diagnosis.
Regional Findings
Radiographic Features by Region
Persistent open fontanelles and sutures with multiple Wormian bones, brachycephaly and frontal/parietal bossing. Midface hypoplasia, underdeveloped paranasal sinuses and a high-arched palate. Bone density is normal.
Absent or hypoplastic clavicles - aplasia is uncommon; partial defects at the junction of the middle and lateral thirds are most frequent. Narrow, bell-shaped thorax with sloping shoulders.
Delayed ossification of the pubis with a wide symphysis, hypoplastic iliac wings, and coxa vara with a varus femoral neck. These are the findings most likely to require orthopaedic intervention.
Scoliosis, kyphosis and spondylolysis may be present. In the hands there is brachydactyly, tapering of the distal phalanges and pseudoepiphyses of the metacarpals.
Dental Imaging
The orthopantomogram (panoramic radiograph) is central to diagnosis and lifelong management. According to PubMed, patients characteristically show retained deciduous teeth, failure of permanent eruption and multiple supernumerary teeth, which together drive the substantial dental burden of the condition (DOI).
Confirmatory Testing
- Molecular genetic testing: Sequence analysis of RUNX2 detects most pathogenic variants; deletion/duplication analysis detects larger rearrangements.
- Skeletal survey: Defines the full extent of skeletal involvement (clavicle, skull, pelvis, spine, hands).
- Family evaluation: Examine and, where appropriate, image parents given variable expressivity.
Clinical Relevance and Differential Diagnosis
Cleidocranial Dysostosis vs Pycnodysostosis
Both share persistent open fontanelles, short stature and clavicular involvement, making this the classic exam comparison. The decisive separator is bone density.
- Cleidocranial Dysostosis
- RUNX2 (CBFA1), autosomal dominant
- Pycnodysostosis
- Cathepsin K (CTSK), autosomal recessive
- Cleidocranial Dysostosis
- Normal (or slightly reduced)
- Pycnodysostosis
- Increased (osteosclerosis)
- Cleidocranial Dysostosis
- Absent
- Pycnodysostosis
- Present (pathognomonic)
- Cleidocranial Dysostosis
- Absent / hypoplastic (hallmark)
- Pycnodysostosis
- Usually mildly hypoplastic/dysplastic
- Cleidocranial Dysostosis
- Supernumerary, unerupted, retained deciduous
- Pycnodysostosis
- Delayed eruption, dental crowding
The shared open fontanelle is a deliberate trap: the bone density on the radiograph (normal here, dense in pycnodysostosis) and the presence of acroosteolysis tell the two apart instantly.
Management
There is no cure and no disease-modifying drug for cleidocranial dysostosis. Management is supportive, anticipatory and multidisciplinary. For the orthopaedic surgeon the key active interventions are correcting symptomatic coxa vara, monitoring the spine, and reassuring families that the dramatic-looking clavicular and cranial findings rarely need surgery. The dental team carries the heaviest long-term treatment load.
Orthopaedic Management
- Clavicles: Usually NO surgery - hypoplasia is well tolerated and rarely symptomatic
- Shoulder girdle: Hypermobility tolerated; physiotherapy for any instability symptoms
- Reassurance: The striking ability to approximate the shoulders is benign
- Coxa vara: Valgus (subtrochanteric) osteotomy for progressive or symptomatic varus
- Spine: Surveillance for scoliosis/kyphosis; brace or operate per standard curve criteria
- Joint laxity: Physiotherapy; address recurrent instability
Coxa vara is the deformity most likely to need surgery. Indications for a valgus proximal femoral osteotomy mirror those for other paediatric coxa vara: a decreasing femoral neck-shaft angle, a steep (vertical) physis (high Hilgenreiner-epiphyseal angle), a symptomatic limp or progressive deformity. The osteotomy realigns the neck, restores the abductor lever arm and corrects the Trendelenburg gait.
Complications
- Coxa vara with limp and abductor weakness
- Scoliosis / kyphosis requiring surveillance or surgery
- Recurrent joint dislocation from ligamentous laxity
- Mild short stature
- Pelvic/obstetric considerations in affected women (narrow pelvis)
- Dental disease: Impaction, malocclusion, caries, abscess
- Obstructive sleep apnoea from midface hypoplasia
- Recurrent otitis media and conductive hearing loss
- Sinusitis from underdeveloped paranasal sinuses
- Cosmetic/psychosocial impact of facial and dental appearance
Affected women may have delayed pubic ossification, a wide symphysis and a relatively narrow pelvis. Combined with a large fetal head (if the fetus is also affected and has delayed cranial ossification), this can complicate delivery - obstetric planning, including consideration of caesarean section, may be warranted.
Guidelines, Registries & Global Practice
Global Epidemiology
Cleidocranial dysostosis is a rare autosomal dominant skeletal dysplasia with an estimated incidence of about 1 per 1 million live births and a roughly equal sex distribution. Around 40% of cases arise de novo, so a negative family history does not exclude the diagnosis. According to PubMed, the genotypic spectrum is dominated by RUNX2 variants in the Runt homology domain, pooled across 453 patients worldwide (DOI). There is no disease-specific international registry; evidence derives from case reports, institutional series and these pooled reviews. Because most surgeons see at most a handful of cases in a career, pattern recognition and referral to a skeletal-dysplasia or craniofacial centre matter more than any single-country pathway.
Why No Society "Guideline" Exists - and What Governs Practice
No orthopaedic society (AAOS, BOA, EFORT, SICOT) publishes a condition-specific guideline for cleidocranial dysostosis given its rarity. Practice is extrapolated from general principles applied to a skeleton with delayed, variable ossification:
- Governing principle
- Valgus proximal femoral osteotomy for symptomatic/progressive varus, as for other paediatric coxa vara
- Source of guidance
- General paediatric-orthopaedic principles
- Governing principle
- Hypoplasia tolerated - surgery almost never indicated
- Source of guidance
- Case-series consensus
- Governing principle
- Standard scoliosis/kyphosis surveillance and treatment thresholds
- Source of guidance
- Scoliosis Research Society principles
- Governing principle
- Staged orthodontic-surgical programme for unerupted/supernumerary teeth
- Source of guidance
- Craniofacial-unit protocols (Jirapinyo 2020)
- Governing principle
- Clinical/radiographic recognition plus RUNX2 molecular confirmation
- Source of guidance
- Clinical genetics standards
- Governing principle
- Autosomal dominant, 50% recurrence, high de novo rate
- Source of guidance
- Clinical genetics standards
Registry and Evidence Notes
- Implant/arthroplasty registries (NJR, AJRR, AOANJRR, SHAR, Norwegian, NZJR) do not capture cleidocranial dysostosis separately; there are no registry-level outcome data specific to this condition.
- Best available synthesis: the 2024 systematic review (453 patients) is the most robust phenotype-genotype resource and the closest thing to evidence-based counselling guidance.
High- vs Limited-Resource Practice Variation
- Well-resourced settings: RUNX2 sequencing, multidisciplinary craniofacial/orthopaedic/dental clinics, image-guided osteotomy and staged orthodontic-surgical dental rehabilitation.
- Limited-resource settings: diagnosis is clinical and radiographic (absent clavicles plus open fontanelles plus dental anomalies); orthopaedic care focuses on the few deformities that are symptomatic (chiefly coxa vara), and basic dental hygiene is high-value and low-cost everywhere.
Genetic Counselling (Universal)
- Autosomal dominant: each child of an affected person has a 50% recurrence risk.
- Approximately 40% are de novo; examine and image apparently unaffected parents given variable expressivity.
- Prenatal or preimplantation genetic testing is feasible once the family's RUNX2 variant is known.
Controversies and Areas of Uncertainty
With an incidence around 1 in a million and no randomised trials, almost all management is extrapolated from case series and general paediatric orthopaedic principles. Examiners reward a candidate who states the principle and then frankly acknowledges the limited evidence base.
There is no trial-level threshold for surgery. Decisions rely on standard coxa vara indicators (falling neck-shaft angle, steep physis, symptomatic limp), extrapolated from other causes of paediatric coxa vara rather than cleidocranial-specific data.
Although RHD missense variants trend toward more severe phenotypes, expressivity within families is so variable that genotype cannot reliably predict an individual's clinical course or counsel precise severity.
Clavicular reconstruction is almost never indicated - hypoplasia is generally asymptomatic. The rare debate concerns thoracic outlet symptoms attributed to clavicular fragments, where evidence is anecdotal.
The ideal sequence and timing of supernumerary-tooth removal and orthodontic traction remains debated; protocols vary between craniofacial units, all based on case-series experience rather than comparative trials.
Why the Teeth Fail to Erupt (and Why There Are Extra Ones)
The topic makes the dental burden central and the cellular card notes 'RUNX2 in the dental follicle', but the body never explains why the teeth fail to erupt or why there are extra ones.
- Eruption needs bone resorption. A tooth erupts only when osteoclasts resorb an eruption pathway through the overlying bone while new bone forms at the base. RUNX2 in the dental follicle drives the signals (such as RANKL and CSF1) that recruit and activate those osteoclasts.
- Why the teeth stay unerupted. With RUNX2 haploinsufficiency the eruption pathway is not properly formed, so permanent teeth fail to erupt and deciduous teeth are retained - the panoramic film shows a mouth full of unerupted teeth.
- Why there are extra teeth. RUNX2 also normally restrains continued tooth formation from the dental lamina, so its deficiency is associated with supernumerary teeth. The two dental hallmarks - failed eruption and extra teeth - therefore both trace back to RUNX2.
Q: Why does RUNX2 haploinsufficiency cause failed eruption and supernumerary teeth?
A: RUNX2 in the dental follicle drives the osteoclast signals (RANKL, CSF1) that resorb an eruption pathway through the overlying bone - so with too little RUNX2 the pathway does not form, permanent teeth fail to erupt and deciduous teeth are retained. RUNX2 also normally restrains continued tooth formation from the dental lamina, so its deficiency is associated with supernumerary teeth. Both dental hallmarks trace to RUNX2 - which is why the dental burden dominates management.
Why the Clavicle and Skull Are the Hallmark Bones
The topic repeatedly says intramembranous bones (clavicle, skull vault) are most affected and that the clavicular defect sits at the middle/lateral-third junction, but never explains why these bones and that spot.
- Membranous bone is the most RUNX2-dose-sensitive. RUNX2 is needed for both ossification types, but intramembranous ossification - the clavicle, the cranial vault and parts of the pubis - depends most on RUNX2 dosage, so a single lost allele hits these membranous bones hardest while the endochondral long bones are relatively spared (hence only mild short stature).
- The clavicle is a special bone. It is the first bone in the body to ossify and forms unusually - an intramembranous shaft from two primary centres, with cartilaginous growth zones at each end.
- Why the defect sits where it does. The two primary centres normally meet near the junction of the middle and lateral thirds; this developmental watershed is exactly where the partial clavicular defect classically appears, which is why complete aplasia is uncommon.
Q: Why are the clavicle and skull the hallmark bones, and why is the clavicular defect at the middle/lateral-third junction?
A: Intramembranous ossification (clavicle, cranial vault, pubis) is the most RUNX2-dose-sensitive, so haploinsufficiency hits these membranous bones hardest while the endochondral long bones are relatively spared (only mild short stature). The clavicle is the first bone to ossify and forms from two primary intramembranous centres (with cartilaginous ends); those centres normally meet near the middle/lateral-third junction - a developmental watershed - which is exactly where the partial defect classically appears (complete aplasia is uncommon).
Viva Practice Scenarios
Practise clinical reasoning and management decisions out loud
“A 4-year-old is referred because she can bring her shoulders together in front of her chest and still has an open fontanelle. Her father has a similar build. What is your diagnosis, and how would you confirm it?”
“You are asked to distinguish cleidocranial dysostosis from pycnodysostosis. Both can have open fontanelles and clavicular changes. How do you tell them apart?”
“A 9-year-old with known cleidocranial dysostosis presents with a progressive limp and a positive Trendelenburg sign. Radiographs show a varus femoral neck. How would you manage this?”
Definition and Key Facts
Molecular Pathogenesis
Cardinal Clinical Features
Radiographic Features
Distinguishing from Pycnodysostosis
Management
Complications
Exam Pearls
Evidence Base
Cleidocranial Dysplasia and RUNX2 - Clinical Phenotype-Genotype Correlation
- RUNX2 (CBFA1) on chromosome 6p21 is the master transcription factor controlling skeletal development
- Two functional isoforms are driven by alternate promoters P1 and P2
- Heterozygous RUNX2 mutations cause cleidocranial dysplasia (autosomal dominant)
- Core features include delayed fontanelle closure, dental abnormalities and hypoplastic clavicles
- Summarises RUNX2 function, mutation types and their phenotypic consequences
The Impact of RUNX2 Gene Variants on Cleidocranial Dysplasia Phenotype: A Systematic Review
- Pooled 569 variants and 453 patients from 103 articles - the largest synthesis to date
- Variants cluster in the Runt homology domain (RHD, 55.5%); roughly half are null and half in-frame
- Missense RHD variants correlate with more severe features (supernumerary teeth, macrocephaly, short ribs, hypoplastic iliac wings)
- In-frame insertions/deletions are associated with fewer CCD features
- Refines phenotype-genotype correlation and informs counselling and management
Cleidocranial Dysplasia: Management of the Multiple Craniofacial and Skeletal Anomalies
- Longitudinal series of 14 patients managed at a single craniofacial unit over four decades
- All had frontal bossing, patent anterior fontanelle, Wormian bones, midface hypoplasia, abnormal dentition and normal intellect
- Obstructive sleep apnoea (11/14), recurrent otitis media and speech/hearing problems were common
- Interventions included ventilation tubes, adenotonsillectomy, orthognathic surgery and cranioplasty
- Demonstrates the essential role of coordinated multidisciplinary management
Cleidocranial Dysplasia: A Clinico-radiographic Spectrum with Differential Diagnosis
- Affects bones derived from both intramembranous and endochondral ossification
- Caused by mutation in CBFA1/RUNX2; autosomal dominant with high penetrance and variable expressivity
- Approximately 40% of cases are sporadic (de novo) with no family history
- Diagnosis is primarily clinical and radiographic, confirmed by molecular genetic testing
- Management requires a multidisciplinary approach including orthopaedic and dental care