Clotting Factor Deficiency and Joint Disease
Hemophilia Classification
Critical Must-Knows
- Hemophilia A: Factor VIII deficiency - most common (85% of cases)
- Hemophilia B: Factor IX deficiency (Christmas disease, 15%)
- Hemophilic Arthropathy: Iron from blood causes synovitis leading to progressive cartilage destruction
- Prophylaxis: Regular factor replacement prevents joint disease - standard of care
- Target Joints: Knee (most common), ankle, elbow - hinge joints more affected
- Perioperative: Raise factor to 100% pre-op, maintain 50-80% post-op, check inhibitor status
Clinical Pearls
- "Factor VIII = Hemophilia A
- "X-linked recessive inheritance
- "Iron toxicity causes arthropathy
- "Prophylaxis prevents joint disease
- "Check inhibitor status preoperatively
Clinical Imaging
Imaging Gallery




Critical Exam Point: Perioperative Factor Management
Orthopaedic surgery in hemophilia requires meticulous factor replacement:
- Pre-operative: Raise factor level to 100%
- Post-operative: Maintain at 50-80% for 7-14 days depending on procedure
- Coordinate with haematology - essential for all procedures
- Check inhibitor status (Bethesda assay) - affects treatment strategy
- Inhibitors present: May need bypassing agents (FEIBA, rFVIIa)
Mnemonics for Exam Recall
ABXHemophilia Types
| A | Factor VIII Most common (85%) - Classic hemophilia |
| B | Factor IX Christmas disease (15%) |
| X | X-linked Recessive - males affected, females carriers |
| A | Factor VIII Most common (85%) - Classic hemophilia |
| B | Factor IX Christmas disease (15%) |
| X | X-linked Recessive - males affected, females carriers |
Hook:ABX - A is VIII, B is IX, X-linked inheritance.
KAETarget Joints: KAE
| K | Knee Most common target joint |
| A | Ankle Second most common |
| E | Elbow Third most common |
| K | Knee Most common target joint |
| A | Ankle Second most common |
| E | Elbow Third most common |
Hook:KAE - Knee, Ankle, Elbow in order of frequency.
CHIPPerioperative Protocol: CHIP
| C | Coordinate haematology Essential multidisciplinary planning |
| H | Hundred percent factor Raise factor to 100% pre-op |
| I | Inhibitor check Bethesda assay before surgery |
| P | Post-op maintain 50-80% factor level for 1-2 weeks |
| C | Coordinate haematology Essential multidisciplinary planning | I | Inhibitor check Bethesda assay before surgery |
| H | Hundred percent factor Raise factor to 100% pre-op | P | Post-op maintain 50-80% factor level for 1-2 weeks |
Hook:CHIP away at perioperative planning.
Overview and Epidemiology
Hemophilia is an X-linked recessive bleeding disorder affecting males.
Types:
- Hemophilia A: Factor VIII deficiency (85% of cases)
- Hemophilia B: Factor IX deficiency (15%, "Christmas disease")
Incidence:
- Hemophilia A: 1 in 5,000 male births
- Hemophilia B: 1 in 30,000 male births
Inheritance:
- X-linked recessive
- Males affected (XY - single X chromosome)
- Females are carriers (usually asymptomatic)
- 30% are new mutations (no family history)
Severity Classification:
- Severe (under 1% factor activity): Spontaneous joint/muscle bleeds
- Moderate (1-5%): Bleeding after minor trauma
- Mild (5-40%): Bleeding after significant trauma/surgery
Pathophysiology
Coagulation Cascade:
- Factor VIII (intrinsic pathway) accelerates Factor X activation
- Factor IX activates Factor X (with Factor VIII as cofactor)
- Deficiency leads to impaired thrombin generation and unstable clots
Hemophilic Arthropathy Development:
- Hemarthrosis: Bleeding into joint space
- Iron deposition: Hemoglobin breakdown releases iron (hemosiderin)
- Synovitis: Iron-induced chronic synovial inflammation
- Enzyme release: Synovial enzymes degrade cartilage
- Cartilage destruction: Progressive chondrolysis
- Secondary changes: Subchondral cysts, osteopenia, osteophytes
- End-stage arthropathy: Joint destruction, contractures, disability
Target Joint Vulnerability:
- Hinge joints (knee, ankle, elbow) more affected than ball-and-socket
- Synovium is highly vascular - susceptible to bleeding
- Repeated bleeds create vicious cycle: bleed to synovitis to more bleeds
Vicious Cycle: Hemarthrosis induces synovitis, which causes synovial proliferation and neovascularisation. The fragile new vessels bleed more easily, leading to recurrent hemarthroses and progressive joint destruction.
Imaging Findings in Hemophilic Arthropathy
Classification
Hemophilia Severity Classification
Classification based on factor activity level determines bleeding phenotype and treatment strategy.
Hemophilia Severity Classification
| Severity | Factor Level | Bleeding Pattern |
|---|---|---|
| Under 1% | Spontaneous joint/muscle bleeds from infancy | |
| 1-5% | Bleeding after minor trauma, occasional spontaneous | |
| 5-40% | Bleeding after significant trauma or surgery |
Most orthopaedic pathology occurs in severe hemophilia due to recurrent spontaneous hemarthroses.
Clinical Presentation
Acute Hemarthrosis
Presentation:
- Acute onset joint pain (often atraumatic)
- Warmth and swelling
- Joint held in position of comfort (usually flexion)
- Limited range of motion
- May have prodromal tingling sensation ("aura")
History:
- May recall minor trauma
- May have been undertreated (missed prophylaxis)
- Frequency of bleeds important for prognosis
Examination:
- Tense effusion
- Increased warmth
- Tenderness on palpation
- Guarding and muscle spasm
- Check for other sites of bleeding
Acute hemarthrosis requires urgent factor replacement to arrest bleeding and minimise joint damage.
Investigations
Laboratory Investigations
Diagnostic:
- Factor VIII assay: Reduced in Hemophilia A
- Factor IX assay: Reduced in Hemophilia B
- APTT: Prolonged (intrinsic pathway)
- PT/INR: Normal (extrinsic pathway intact)
- Bleeding time: Normal (platelet function intact)
Preoperative Workup:
- Inhibitor screen (Bethesda assay): Critical - antibodies to factor
- Factor level and recovery study
- Blood group and crossmatch
- Hepatitis B, C and HIV status (historical transfusion risk)
- Full blood count
- Liver function (may have hepatitis-related liver disease)
Monitoring:
- Factor levels during treatment
- Trough levels if on prophylaxis
Inhibitor development occurs in 25-30% of severe Hemophilia A patients and significantly complicates treatment.
Hemophilia A vs B:
Hemophilia A vs B
| Feature | Hemophilia A | Hemophilia B |
|---|---|---|
| Factor VIII | Factor IX | |
| 85% of cases | 15% of cases | |
| Classic hemophilia | Christmas disease | |
| Factor VIII concentrate | Factor IX concentrate | |
| 25-30% in severe | 3-5% in severe |
Differential Diagnosis
A swollen, painful joint in a young male is not always hemophilic hemarthrosis. The key orthopaedic discriminator is the bleeding history and coagulation profile.
Differential Diagnosis of the Acute Painful Swollen Joint
| Condition | Distinguishing Features | Coagulation / Key Test |
|---|---|---|
| Recurrent atraumatic bleeds, target joint, prodromal aura, known family history | Prolonged APTT, normal PT, reduced FVIII or FIX | |
| Fever, systemic upset, exquisite pain on micro-movement; can coexist with hemophilia | Raised CRP/WCC, aspirate for Gram stain and culture (with factor cover) | |
| Mucocutaneous bleeding predominates, hemarthrosis rare except severe type 3 | Reduced vWF antigen/activity, variable FVIII | |
| Symmetrical or polyarticular, morning stiffness, no bleeding history | Normal coagulation, raised inflammatory markers, ANA | |
| Single joint, recurrent blood-stained effusions, no clotting defect | MRI low-signal hemosiderin (blooming), normal coagulation | |
| Clear injury mechanism, rapid effusion, normal clotting | Normal APTT/PT, MRI shows structural injury |
Management
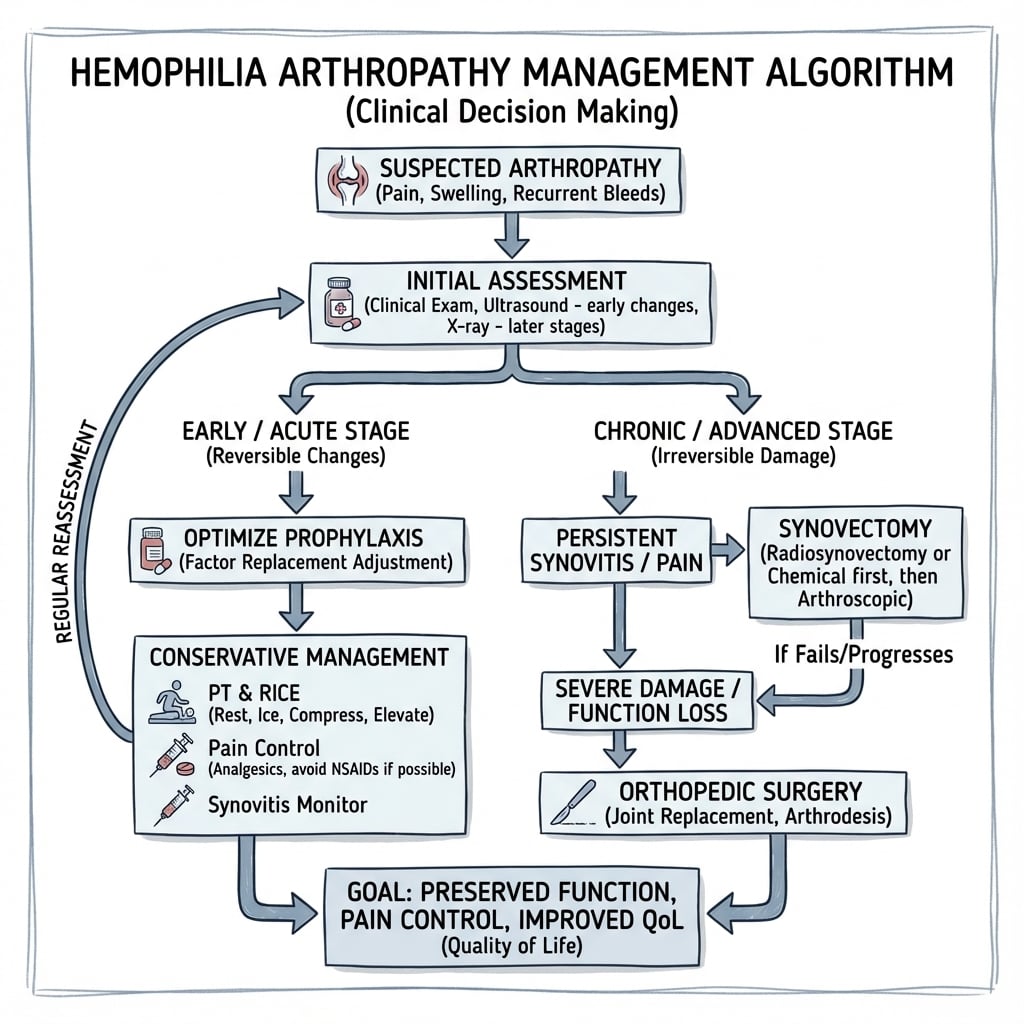
Primary Prevention (Prophylaxis)
Standard of Care:
- Regular factor replacement prevents hemarthroses
- Start before or soon after first joint bleed
- Aim: Maintain trough level above 1-5%
Prophylaxis Regimens:
- Hemophilia A: Factor VIII 25-40 IU/kg every other day or 3x/week
- Hemophilia B: Factor IX 25-40 IU/kg twice weekly
Evidence (Landmark Joint Outcome Study):
- Manco-Johnson et al, NEJM 2007 (severe Hemophilia A, boys under 30 months)
- Prophylaxis vs enhanced episodic treatment
- Prophylaxis group: 93% index joints normal on MRI at age 6
- Episodic group: 55% normal (relative risk of damage 6.1)
- Prophylaxis is now standard of care
Extended Half-Life and Non-Factor Products:
- EHL FVIII/IX (Fc or albumin fusion, PEGylation) reduce dosing frequency and raise troughs
- Emicizumab (subcutaneous bispecific antibody) for Hemophilia A with or without inhibitors - HAVEN 3 showed 96-97% bleed reduction
- Improved adherence and quality of life
Early prophylaxis prevents the development of hemophilic arthropathy.
Complications
Orthopaedic Complications:
- Progressive joint destruction (arthropathy)
- Fixed flexion contractures
- Angular deformity (valgus/varus)
- Muscle wasting and weakness
- Limb length discrepancy
- Pseudotumours (rare - encapsulated haematomas in soft tissue/bone)
Surgical Complications:
- Intraoperative bleeding
- Postoperative haematoma
- Infection (higher rate than general population)
- Wound healing problems
- Stiffness (especially knee)
- DVT/PE (paradoxical - still occurs)
Treatment Complications:
- Inhibitor development: Antibodies to factor (25-30% Hemophilia A)
- Transfusion-transmitted infections (historical - HCV, HIV)
- Allergic reactions to factor concentrates
- Central venous catheter complications (if port present)
Pseudotumour:
- Rare but serious
- Encapsulated haematoma in muscle or bone
- Progressive enlargement if untreated
- May erode bone
- Treatment: Factor replacement, surgery if large
Controversies & Areas of Uncertainty
- Optimal prophylaxis target. Traditional regimens aim for troughs above 1%, but breakthrough bleeds still occur at this level. There is ongoing debate about individualised, higher-trough or pharmacokinetic-guided dosing versus fixed regimens, balanced against cost and venous access.
- Role of emicizumab in non-inhibitor patients. Non-factor therapy dramatically lowers bleeding rates, but long-term joint-protection data and its interaction with breakthrough bleeds requiring factor or bypassing agents are still maturing.
- Joint aspiration in acute hemarthrosis. Aspiration may relieve a tense effusion and reduce iron load, but carries infection and re-bleed risk; indications remain debated and it should never precede factor cover.
- Timing and choice of synovectomy. Radiosynovectomy, arthroscopic and open synovectomy all reduce bleeding, but comparative long-term superiority and the threshold for intervention are not firmly established; radionuclide availability drives practice regionally.
- Implant choice in arthroplasty. The balance between cemented vs cementless fixation and constrained vs less-constrained knee implants in young, often osteopenic hemophilic patients with deformity is unresolved.
- VTE prophylaxis after major surgery. Hemophilia is not fully protective against venous thromboembolism, especially with factor correction; whether and how to give chemical thromboprophylaxis perioperatively is genuinely uncertain and individualised with haematology.
- Gene therapy durability. AAV-mediated factor gene transfer can achieve sustained factor expression, but durability, hepatotoxicity, immune responses and long-term joint outcomes remain under active study.
Outcomes
Prophylaxis Era Outcomes:
- Joint disease markedly reduced
- Joint Outcome Study: 93% index joints normal on MRI with prophylaxis at age 6
- Life expectancy approaching normal
- Quality of life dramatically improved
Without Prophylaxis (Historical):
- Severe arthropathy by adolescence
- Wheelchair dependence common
- Multiple joint replacements
- Significant disability
Arthroplasty Outcomes:
- Pain relief: 85-90% satisfied
- Function improvement: Significant
- Survivorship: 85-90% at 10 years
- Complications higher than general population
- Revision rate higher
Prognostic Factors:
- Severity of hemophilia (severe worse)
- Adherence to prophylaxis (critical)
- Inhibitor status (inhibitors worsen prognosis)
- Age at first bleed (earlier worse)
- Number of target joints
Evidence Base
- RCT of 65 boys under 30 months with severe Hemophilia A: prophylactic recombinant FVIII vs enhanced episodic therapy
- At age 6, normal index-joint structure on MRI in 93% (prophylaxis) vs 55% (episodic), P=0.006
- Relative risk of MRI joint damage with episodic therapy 6.1 (95% CI 1.5-24.4)
- Joint and total hemorrhage rates significantly lower with prophylaxis
- Cohort of 76 patients with severe hemophilia, median follow-up to age 19
- Median age at first joint bleed 2.2 years; prophylaxis started median age 6
- Pettersson score was 8% higher (95% CI 1-16%) for every year prophylaxis was postponed after the first joint bleed
- Effect independent of age at scoring and prophylactic dose
- Phase 3 RCT of subcutaneous emicizumab in 152 patients with hemophilia A WITHOUT inhibitors
- Annualised bleeding rate 1.5 (weekly) and 1.3 (fortnightly) vs 38.2 with no prophylaxis - 96-97% reduction
- Intra-individual comparison: 68% lower bleeding than prior FVIII prophylaxis
- No thrombotic events or new FVIII inhibitors; main adverse event mild injection-site reaction
Viva Scenarios
Use these scenarios to practise clinical reasoning and management decisions
Acute Hemarthrosis
"12-year-old boy with severe Hemophilia A presents with acute left knee swelling and pain after playing soccer. He is on regular prophylaxis but missed yesterday's dose."
This is an acute hemarthrosis in a known hemophiliac. The missed prophylaxis dose likely contributed to bleeding vulnerability.
Immediate management: Factor VIII replacement to raise level to 40-60%. Do not wait for investigations - treat first. Repeat doses may be needed (usually 1-3).
Supportive care (RICE): Rest (brief immobilisation), Ice, Compression, Elevation. Joint aspiration is controversial - consider only if very tense effusion with factor cover due to infection risk.
Early mobilisation: Once bleeding controlled, begin physiotherapy to maintain ROM. Prolonged immobilisation causes contractures.
Review prophylaxis: Discuss importance of adherence. Consider if regimen is adequate (may need dose adjustment).
Severe Arthropathy - TKR Planning
"35-year-old man with severe Hemophilia A, no inhibitors, presents with severe right knee arthropathy. Fixed 30-degree flexion contracture, valgus deformity, pain limiting mobility. Conservative management has failed."
This patient has end-stage hemophilic arthropathy of the knee and is a candidate for total knee replacement.
Preoperative workup (CHIP protocol):
- Coordinate with haematology - essential, cannot proceed without them
- Hundred percent factor VIII pre-operatively
- Inhibitor screen (Bethesda assay) - critical, changes management entirely
- Post-op plan: Maintain factor 50-80% for 10-14 days
Surgical considerations: May need constrained implant due to ligament laxity. Address fixed flexion contracture (posterior release). Expect higher blood loss and complication rate than standard TKR. Meticulous haemostasis intraoperatively.
Counselling: Higher complication rates (infection 5-10%, stiffness common), but good pain relief expected. Results improving with modern protocols.
Recurrent Hemarthroses Despite Prophylaxis
"8-year-old boy with moderate Hemophilia A has recurrent right knee hemarthroses despite prophylaxis. X-ray shows early arthropathy with widened intercondylar notch. What do you recommend?"
This patient has a target joint with chronic synovitis leading to recurrent bleeds despite prophylaxis. The widened intercondylar notch is a classic radiographic sign of hemophilic knee arthropathy.
Assessment:
- Confirm prophylaxis compliance and adequacy
- Check trough factor levels - may need dose increase
- MRI to assess synovitis severity
- Consider inhibitor screen
Management options:
- Optimise prophylaxis: Increase dose or frequency
- Radiosynoviorthesis: Injection of radioactive isotope (P-32, Y-90). 85% reduction in bleeding episodes. Can repeat if needed. Low complication rate.
- Arthroscopic synovectomy: If radiosynovectomy fails or unavailable
Radiosynoviorthesis is effective and minimally invasive - first-line after optimising prophylaxis.
Inhibitor Patient
"20-year-old with severe Hemophilia A needs elbow synovectomy for recurrent hemarthroses. Inhibitor screen shows Bethesda titre of 12 BU. How does this change your approach?"
This patient has a high-titre inhibitor (greater than 5 BU), which significantly complicates management. Standard Factor VIII replacement will be ineffective as antibodies neutralise the factor.
Management options:
- Bypassing agents:
- FEIBA (Factor Eight Inhibitor Bypassing Activity) - activated prothrombin complex concentrate
- Recombinant Factor VIIa (rFVIIa, NovoSeven)
- These bypass the need for Factor VIII in the coagulation cascade
- Monitoring is more difficult (cannot use factor levels)
Considerations:
- Higher bleeding risk than non-inhibitor patients
- More challenging perioperative management
- Expert haematology input essential
- May need to consider immune tolerance induction for long-term
High-titre inhibitor patients should be managed in specialist centres with haemophilia expertise.
Guidelines, Registries & Global Practice
Global Epidemiology:
- Hemophilia A affects approximately 1 in 5,000 male births; Hemophilia B approximately 1 in 30,000 male births
- Hemophilia A accounts for roughly 80-85% of cases worldwide; Hemophilia B for 15-20%
- The World Federation of Hemophilia (WFH) Annual Global Survey identifies over 200,000 people with hemophilia, but case identification remains far below expected prevalence in low-income regions
- Around 30% of cases arise from de novo mutations with no family history
Side-by-Side Guidelines:
Major Society Guidance on Hemophilia Care
| Body | Emphasis | Key Recommendation |
|---|---|---|
| Comprehensive global standard | Prophylaxis for all severe hemophilia; integrated multidisciplinary care including musculoskeletal assessment | |
| Treatment products and prophylaxis | Early prophylaxis from first joint bleed or young age; supports EHL and non-factor agents | |
| National protocols and inhibitor management | Standardised perioperative factor dosing and structured inhibitor surveillance | |
| Surgical decision-making | Mandatory haematology co-management; confirmed factor/inhibitor plan before any elective procedure |
Registries & Surveillance:
- WFH World Bleeding Disorders Registry collects longitudinal outcome and treatment data across countries
- National arthroplasty registries (NJR, AJRR, AOANJRR, Swedish/Norwegian) capture small but informative volumes of hemophilic joint replacements - consistently showing higher revision and infection rates than osteoarthritis
- Patient-reported and joint-health tools (Haemophilia Joint Health Score, HAL, FISH) increasingly standardised for global comparison
High- vs Limited-Resource Practice Variation:
- High-resource settings: primary prophylaxis from infancy, EHL and non-factor agents (emicizumab), point-of-care ultrasound, elective arthroplasty for end-stage disease
- Limited-resource settings: factor supply is the dominant constraint; care is often on-demand or low-dose ("intermediate-dose") prophylaxis, with later presentation and more advanced arthropathy
- Lower-cost radionuclides (e.g. rhenium-188) and synovectomy are emphasised where repeated arthroplasty is not feasible
- Gene therapy (adeno-associated virus FVIII/FIX transfer) is emerging but currently limited to high-resource, highly selected populations
HEMOPHILIA ORTHOPAEDIC
Clinical summary
TYPES
- •Hemophilia A: Factor VIII (85%)
- •Hemophilia B: Factor IX (15%)
- •X-linked recessive
- •Severe: under 1% factor
TARGET JOINTS (KAE)
- •Knee most common
- •Ankle second
- •Elbow third
- •Hinge joints vulnerable
PREVENTION
- •Prophylaxis standard of care
- •JOS: 93% joints normal on MRI
- •Start soon after first joint bleed
- •Trough above 1-5%
PERIOPERATIVE (CHIP)
- •Coordinate haematology
- •Hundred percent factor pre-op
- •Inhibitor check (Bethesda)
- •Post-op 50-80% for 2 weeks