Multinucleated Bone-Resorbing Cells | RANKL-RANK Pathway | Ruffled Border
- Osteoclasts are multinucleated (10-100 nuclei) cells derived from hematopoietic stem cells
- RANKL-RANK pathway is essential for osteoclast differentiation and activation
- Ruffled border creates acidic microenvironment (pH 4.5) to dissolve hydroxyapatite
- Cathepsin K and matrix metalloproteinases degrade organic bone matrix
- Osteoprotegerin (OPG) acts as decoy receptor, inhibiting RANKL-RANK binding
- “Howship lacuna is the resorption pit created by active osteoclasts
- “RANK mutations cause osteopetrosis (marble bone disease)
- “Bisphosphonates induce osteoclast apoptosis by inhibiting farnesyl pyrophosphate synthase
- “Denosumab is monoclonal antibody against RANKL, preventing RANK binding
Hematopoietic lineage. Osteoclasts derive from monocyte-macrophage precursors in bone marrow, NOT from mesenchymal stem cells like osteoblasts.
Master regulatory pathway. RANKL (from osteoblasts/stromal cells) binds RANK (on osteoclast precursors); OPG acts as decoy receptor.
Specialized membrane. Creates sealed acidic compartment (pH 4.5) via H+ ATPase proton pumps, dissolving mineral phase.
Therapeutic interventions. Bisphosphonates, denosumab, and calcitonin all target osteoclast activity in osteoporosis.
Overview and Cell Biology
Osteoclasts are the ONLY cells capable of resorbing mineralized bone. They are multinucleated giant cells (10-100 nuclei) derived from hematopoietic monocyte-macrophage lineage, making them fundamentally different from bone-forming osteoblasts (mesenchymal origin). This dual-origin system is essential for bone remodeling balance.
- Size: 100-150 μm diameter
- Nuclei: 10-100 per cell (from fusion)
- Appearance: Multinucleated giant cell
- Lifespan: Approximately 2 weeks
- Location: Howship lacunae (resorption pits)
- Stem cell: Hematopoietic stem cell
- Lineage: Monocyte-macrophage pathway
- Precursors: Circulating monocytes
- Fusion: Multinucleation required for function
- Relation: Share origin with macrophages
Concepts and Molecular Pathways
Key Osteoclast Concepts:
- RANK/RANKL/OPG Axis: RANKL activates RANK receptor on precursors; OPG is decoy receptor
- Ruffled Border: Specialized membrane creating acidic microenvironment (pH 4.5)
- Sealing Zone: Actin ring isolates resorption compartment
- Therapeutic Targets: Bisphosphonates (apoptosis), denosumab (RANKL inhibition)
Osteoclast Differentiation and Activation
Osteoclastogenesis Pathway
Hematopoietic stem cells in bone marrow differentiate into monocyte-macrophage precursors. M-CSF (macrophage colony-stimulating factor) is essential for precursor survival and proliferation.
RANKL (receptor activator of nuclear factor kappa-B ligand) produced by osteoblasts and stromal cells binds to RANK receptors on precursors. This is the critical commitment step.
Multinucleation occurs as mononuclear precursors fuse to form giant cells with 10-100 nuclei. Dendritic cell-specific transmembrane protein (DC-STAMP) mediates fusion.
Ruffled border formation and sealing zone development. Cell attaches to bone via αvβ3 integrin, creating sealed resorption compartment.
Bone dissolution via acidification (dissolves mineral) and enzyme release (degrades matrix). Lasts hours to days.
Programmed cell death after resorption cycle complete. Triggered by loss of RANKL signal or OPG inhibition.
The balance between RANKL (activator) and OPG (osteoprotegerin, decoy receptor) determines osteoclast number and activity. OPG is produced by osteoblasts and binds RANKL, preventing RANK activation. The RANKL:OPG ratio is the key determinant of bone resorption rate. This is the target of denosumab therapy.
Molecular Mechanisms of Bone Resorption
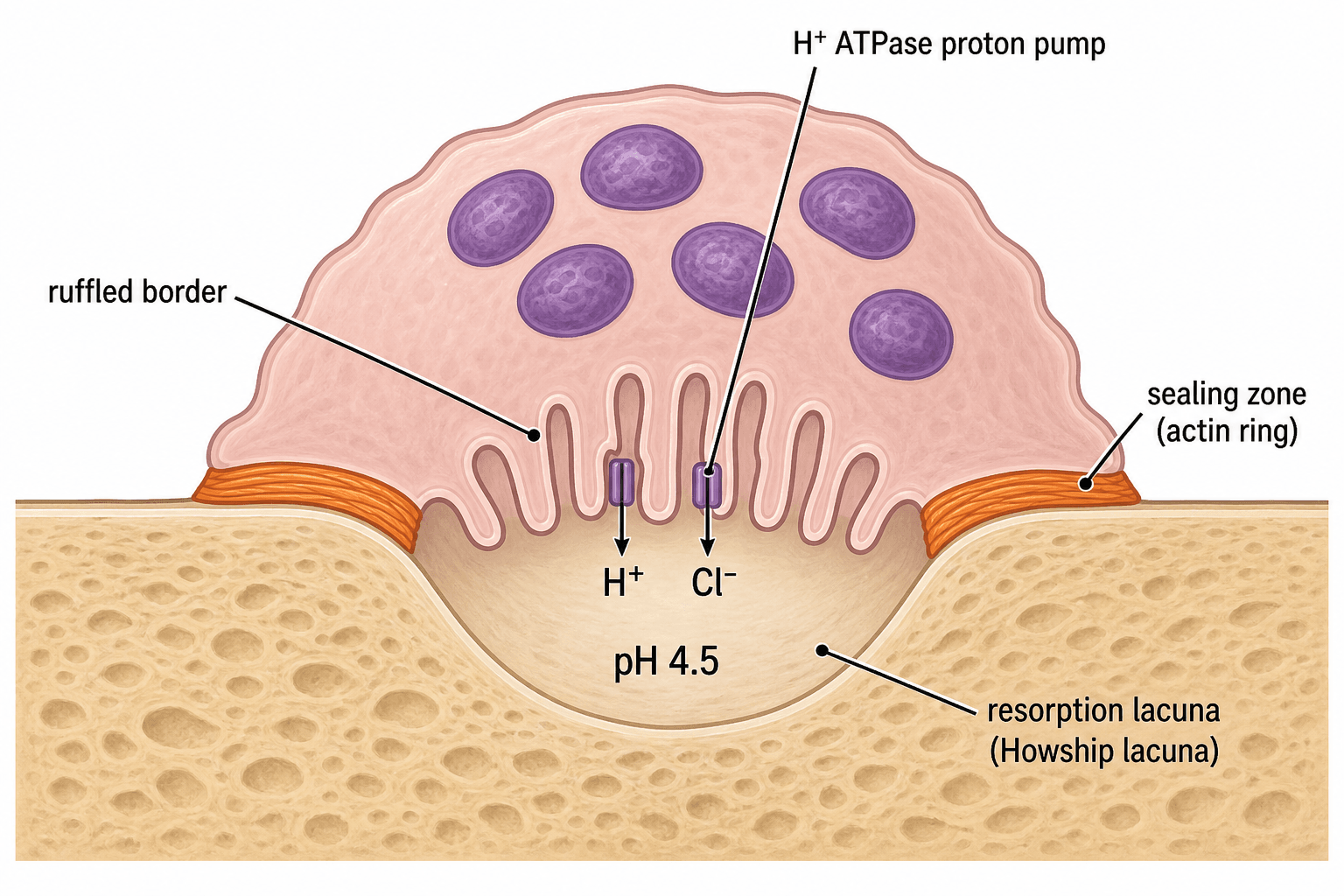
Mineral Phase Dissolution
H+ ATPase (V-type) in ruffled border membrane actively pumps protons into resorption lacuna, creating pH 4.5 environment.
ClC-7 chloride channels maintain electroneutrality by transporting Cl- ions alongside H+ ions.
Result: Hydroxyapatite crystals dissolve in acidic environment, releasing calcium and phosphate.
Carbonic anhydrase II enzyme generates H+ ions from CO2 + H2O inside osteoclast. Mutations cause osteopetrosis with renal tubular acidosis.
Regulation of Osteoclast Activity
- Source
- Osteoblasts/stromal cells
- Effect
- Stimulates +++
- Mechanism
- Binds RANK, activates NFκB
- Source
- Stromal cells/osteoblasts
- Effect
- Stimulates ++
- Mechanism
- Precursor survival/proliferation
- Source
- Osteoblasts
- Effect
- Inhibits ---
- Mechanism
- Decoy receptor for RANKL
- Source
- Parathyroid gland
- Effect
- Stimulates (indirect)
- Mechanism
- Increases RANKL expression
- Source
- Kidney (activated)
- Effect
- Stimulates (indirect)
- Mechanism
- Increases RANKL expression
- Source
- Gonads
- Effect
- Inhibits
- Mechanism
- Suppresses RANKL, increases OPG
- Source
- Thyroid C-cells
- Effect
- Inhibits
- Mechanism
- Direct receptor on osteoclast
Postmenopausal estrogen deficiency increases RANKL and decreases OPG production, shifting the RANKL:OPG ratio toward bone resorption. This explains accelerated bone loss in postmenopausal women and the efficacy of estrogen replacement therapy.
Clinical Applications and Pathology
Pathological States:
- Osteoporosis (postmenopausal, steroid-induced)
- Paget disease (abnormal osteoclasts)
- Hyperparathyroidism
- Multiple myeloma
- Bone metastases
Pathological States:
- Osteopetrosis (RANK/RANKL/ClC-7 mutations)
- Pycnodysostosis (cathepsin K deficiency)
- Bisphosphonate therapy (excessive)
- Carbonic anhydrase II deficiency
Osteopetrosis results from osteoclast dysfunction due to mutations in RANK, RANKL, carbonic anhydrase II, or ClC-7 chloride channel. Results in dense sclerotic bone (marble bone) that is paradoxically fragile, with obliteration of marrow spaces causing cytopenias. Severe forms require hematopoietic stem cell transplantation to provide functional osteoclast precursors.
Bone Resorption Markers (CTX, NTX, TRAP-5b)
The enzyme table and cheat sheet call TRAP "a serum marker of resorption", and Viva 3 asks which marker reflects osteoclast number — here are the resorption markers and what each actually measures.
- What it is
- A collagen breakdown fragment released when osteoclasts degrade matrix
- What it reflects
- Osteoclast activity (resorption rate); the preferred serum resorption marker for monitoring antiresorptive response/adherence (drawn fasting; some use it to gauge ONJ risk)
- What it is
- A collagen breakdown fragment (urine or serum)
- What it reflects
- Osteoclast activity; an older resorption marker
- What it is
- An enzyme secreted by osteoclasts themselves
- What it reflects
- Osteoclast number (not just activity), and it is independent of renal function and food intake
Resorption markers fall rapidly when an antiresorptive is started (a CTX drop confirms response/compliance) and surge when denosumab is stopped — the biochemical signature of the rebound.
The osteopetrosis answer: in the common osteoclast-RICH forms (TCIRG1, CLCN7, carbonic anhydrase II — osteoclasts present but unable to resorb) TRAP-5b is HIGH because there are many osteoclasts; in the osteoclast-POOR RANK/RANKL forms it is LOW because osteoclasts fail to form. So TRAP-5b separates the two osteopetrosis subtypes and predicts whether haematopoietic stem cell transplantation can work. The bone-FORMATION markers (P1NP, osteocalcin, bone-specific ALP) and the remodeling cycle itself are developed in the osteoblasts-bone-formation and bone-remodeling topics.
Q: Which resorption marker reflects osteoclast NUMBER rather than activity, and why does it matter in osteopetrosis? A: TRAP-5b is secreted by osteoclasts and tracks their number; CTX/NTX are collagen fragments that track resorptive activity. In osteopetrosis TRAP-5b is high in the osteoclast-rich acidification-defect forms but low in the osteoclast-poor RANK/RANKL forms — distinguishing the subtypes that will or will not respond to HSCT.
Carbonic Anhydrase II Deficiency (Sly Syndrome)
The acidification tab notes that carbonic anhydrase II (CA-II) deficiency causes osteopetrosis "with renal tubular acidosis", and Viva 3 asks why — here is the mechanism. CA-II catalyses CO2 + H2O into H+ and bicarbonate, generating the very protons the osteoclast's H+-ATPase pumps into the resorption lacuna. The same enzyme is expressed in the renal tubule and the brain, so its autosomal-recessive loss produces a characteristic triad:
- Osteopetrosis — without CA-II-generated protons the osteoclast cannot acidify the lacuna or dissolve hydroxyapatite, so dense bone accumulates (the osteoclasts are present but non-functional).
- Renal tubular acidosis — CA-II is required for renal tubular acid-base handling, so its loss impairs both proximal bicarbonate reclamation and distal urinary acidification (a mixed, "type 3" RTA), producing a metabolic acidosis.
- Cerebral calcification (typically the basal ganglia) with developmental delay.
This renal-and-cerebral phenotype is what distinguishes CA-II deficiency from the TCIRG1 and CLCN7 acidification defects (which give isolated osteopetrosis); CA-II deficiency is also a milder, often later-presenting form. The detailed osteopetrosis subtypes and the role of HSCT are developed in the osteopetrosis topic.
Q: Why does carbonic anhydrase II deficiency cause renal tubular acidosis as well as osteopetrosis? A: CA-II generates H+ (from CO2 and water) for both the osteoclast ruffled border and the renal tubule. Losing it blocks osteoclast acidification (osteopetrosis) AND renal tubular acid handling — impaired proximal bicarbonate reclamation plus distal urinary acidification — giving a mixed (type 3) RTA, completed by basal-ganglia cerebral calcification (the Sly triad).
Differential Diagnosis: Osteoclast-Driven Bone Disorders
When osteoclast number or function is abnormal, several conditions can present with overlapping radiographic or biochemical features. Distinguishing them rests on osteoclast biology.
- Osteoclast Defect
- Excess resorption (high RANKL:OPG)
- Bone Density
- Reduced
- Key Distinguisher
- Low BMD, fragility fractures, high bone turnover markers
- Osteoclast Defect
- Giant, hyperactive, viral-inclusion osteoclasts
- Bone Density
- Mixed lytic/sclerotic
- Key Distinguisher
- Markedly raised ALP, mosaic lamellar bone, bone pain/deformity
- Osteoclast Defect
- Present but cannot resorb (TCIRG1, CLCN7, CA-II)
- Bone Density
- Markedly increased
- Key Distinguisher
- Dense brittle bone, marrow failure, cranial nerve palsies
- Osteoclast Defect
- Osteoclast-poor (fail to form)
- Bone Density
- Markedly increased
- Key Distinguisher
- Few/absent osteoclasts; HSCT ineffective for RANKL form
- Osteoclast Defect
- Cathepsin K deficiency
- Bone Density
- Increased
- Key Distinguisher
- Acro-osteolysis, short stature, retained collagen matrix
- Osteoclast Defect
- PTH-driven focal hyperresorption
- Bone Density
- Focal lytic
- Key Distinguisher
- Raised PTH/calcium, subperiosteal resorption, giant-cell lesion
Guidelines, Registries & Global Practice
Global Epidemiology
- Osteoporosis affects an estimated 500 million people worldwide; roughly 1 in 3 women and 1 in 5 men over 50 will sustain a fragility fracture.
- Over 8.9 million osteoporotic fractures occur globally each year (a fragility fracture roughly every 3 seconds), with hip fractures projected to rise sharply in Asia as populations age.
- Paget disease shows marked geographic variation — historically common in the UK and populations of British descent, and declining in prevalence in recent decades.
- Infantile (malignant) osteopetrosis has an incidence of approximately 1 in 250,000 births; the autosomal dominant adult form is far more common at around 1 in 20,000.
Major Guidelines, Side by Side
- First-line
- Oral/IV bisphosphonate; denosumab or anabolic for very high risk
- Denosumab Stance
- First-line option in high/very-high risk
- Discontinuation Caveat
- Never stop without follow-on anti-resorptive
- First-line
- Oral bisphosphonate (alendronate/risedronate)
- Denosumab Stance
- Where bisphosphonate unsuitable or higher risk
- Discontinuation Caveat
- Mandatory bisphosphonate bridge on stopping
- First-line
- Bisphosphonate; sequential anabolic-then-antiresorptive in severe disease
- Denosumab Stance
- Recognised potent anti-resorptive
- Discontinuation Caveat
- Structured transition to avoid rebound
- First-line
- Treat above country-specific intervention threshold
- Denosumab Stance
- Reserved per fracture-risk tier
- Discontinuation Caveat
- Uniform rebound warning
- Consensus across societies: nitrogen-containing bisphosphonates remain first-line; denosumab is a potent alternative but must never be stopped abruptly; teriparatide/romosozumab (anabolic) are favoured first for very-high-risk patients, followed by an anti-resorptive to consolidate gains.
- Anti-resorptive "drug holidays" apply to bisphosphonates (which persist in bone for years) but are explicitly contraindicated for denosumab.
Registry and Resource-Setting Notes
- Arthroplasty registries (NJR, AJRR, AOANJRR, SHAR, Norwegian, NZJR) track periprosthetic fractures, which are influenced by underlying bone quality and anti-resorptive status; bisphosphonate use around arthroplasty and its effect on aseptic loosening and revision is an area of ongoing analysis.
- In well-resourced settings, severe infantile osteopetrosis is managed with haematopoietic stem cell transplantation and bone-density-targeted pharmacotherapy is guided by DXA and bone turnover markers.
- In limited-resource settings, DXA access, the cost of denosumab and anabolic agents, and HSCT availability are major constraints; generic oral bisphosphonates and FRAX (which can be calculated without BMD) are the practical mainstays.
Controversies and Areas of Uncertainty
Odanacatib reduced fractures in the LOFT trial but was withdrawn in 2016 after a signal of increased stroke risk. Whether selective cathepsin K inhibition can be achieved without off-target cardiovascular or cutaneous effects remains unresolved.
The rebound phenomenon is established, but the ideal bridging regimen (which bisphosphonate, oral vs IV, timing relative to the missed dose, and duration) is not defined by prospective trials and varies between guidelines.
Balancing atypical femoral fracture and osteonecrosis-of-jaw risk (rising with cumulative exposure) against rebound fracture risk on stopping is contentious. Holidays are reasonable for bisphosphonates but are explicitly unsafe for denosumab.
Beyond resorption, osteoclasts secrete "clastokines" that couple resorption to formation. Whether anabolic benefit can be preserved while suppressing resorption (the basis of interest in coupling-sparing agents) is an active question.
Treatment sequence matters: giving an anti-resorptive (denosumab/bisphosphonate) before the anabolic teriparatide blunts the anabolic response, whereas the reverse sequence (anabolic first, then anti-resorptive to consolidate gains) is preferred. This reflects the dependence of teriparatide on a "remodelling space" created by active osteoclasts.
Pharmacological Targeting of Osteoclasts
Mechanism of Action
Nitrogen-containing bisphosphonates (alendronate, risedronate, zoledronic acid) inhibit farnesyl pyrophosphate synthase in the mevalonate pathway, preventing prenylation of small GTPases essential for osteoclast function.
Bisphosphonate Action
Bisphosphonates bind hydroxyapatite with high affinity, becoming incorporated into bone matrix.
During resorption, osteoclasts endocytose bisphosphonate-containing bone.
Intracellular bisphosphonate inhibits farnesyl pyrophosphate synthase, disrupting GTPase signaling.
Loss of functional GTPases triggers osteoclast apoptosis, reducing bone resorption.
Adverse effects: Osteonecrosis of jaw (rare), atypical femoral fractures (with prolonged use greater than 5 years).
MCQ Practice Points
Q: Osteoclasts are derived from which cell lineage? A: Hematopoietic monocyte-macrophage lineage - NOT mesenchymal. This is why bone marrow transplantation can cure some forms of osteopetrosis by providing functional osteoclast precursors.
Q: What is the receptor for RANKL on osteoclast precursors? A: RANK (receptor activator of nuclear factor kappa-B). Activation leads to NFκB signaling and osteoclastogenesis. Mutations cause osteopetrosis.
Q: What is the major collagenase enzyme secreted by osteoclasts? A: Cathepsin K - accounts for the majority of type I collagen degradation. Functions optimally at acidic pH. Deficiency causes pycnodysostosis.
Q: How do nitrogen-containing bisphosphonates cause osteoclast apoptosis? A: Inhibit farnesyl pyrophosphate synthase in the mevalonate pathway, preventing prenylation of small GTPases required for osteoclast function and survival.
At a Glance
Osteoclasts are multinucleated (10-100 nuclei) bone-resorbing cells derived from the hematopoietic monocyte-macrophage lineage, fundamentally distinct from osteoblasts which arise from mesenchymal stem cells. The RANKL-RANK-OPG axis serves as the master regulatory pathway: RANKL from osteoblasts/stromal cells binds RANK on osteoclast precursors to drive differentiation, while osteoprotegerin (OPG) acts as a decoy receptor to inhibit this interaction. Active osteoclasts form a specialized ruffled border membrane that creates a sealed acidic microenvironment (pH 4.5) via H+-ATPase proton pumps to dissolve hydroxyapatite, with cathepsin K and matrix metalloproteinases degrading the organic matrix. This pathway is therapeutically targeted by bisphosphonates (induce osteoclast apoptosis via FPP synthase inhibition) and denosumab (RANKL monoclonal antibody) in osteoporosis management.
RANKRANKL-RANK Pathway Components
Hook:RANK kills bone - the receptor-activator pathway that drives osteoclast formation!
RSCBOsteoclast Functional Zones
Hook:RSCB - Ruffled Sealing Creates Breakdown of bone architecture!

Exam Viva Scenarios
Practise clinical reasoning and management decisions out loud
“The examiner shows you a histological image of bone tissue with multinucleated cells in Howship lacunae. Describe what you see and explain the cell function.”
“Explain the molecular regulation of osteoclast differentiation and how this relates to osteoporosis treatment.”
“A child presents with dense, sclerotic bones on radiograph, anaemia, recurrent infections and cranial nerve palsies. Bone biopsy shows abundant but dysfunctional osteoclasts. Explain the pathophysiology and how this contrasts with pycnodysostosis, and outline management.”
Key Cell Biology
- Multinucleated (10-100 nuclei) from hematopoietic monocyte lineage
- Lifespan approximately 2 weeks
- Located in Howship lacunae (resorption pits)
- Ruffled border membrane facing bone, basolateral for transcytosis
RANKL-RANK-OPG Axis
- RANKL (osteoblast) + RANK (osteoclast precursor) = activation
- OPG = decoy receptor, blocks RANKL-RANK binding
- RANKL:OPG ratio determines resorption rate
- M-CSF required for precursor survival
Resorption Mechanism
- H+ ATPase pumps create pH 4.5 in sealed lacuna
- Acidic pH dissolves hydroxyapatite mineral
- Cathepsin K degrades type I collagen matrix
- Sealing zone (actin ring) maintains isolation
Pharmacological Targets
- Bisphosphonates: inhibit farnesyl pyrophosphate synthase, induce apoptosis
- Denosumab: anti-RANKL antibody, prevents RANK binding
- Calcitonin: direct osteoclast receptor, rapid inhibition
- Denosumab discontinuation causes rebound resorption
Clinical Conditions
- Osteopetrosis: RANK/RANKL/ClC-7/CA-II mutations, dense fragile bone
- Pycnodysostosis: cathepsin K deficiency
- Postmenopausal osteoporosis: increased RANKL:OPG ratio
- Paget disease: abnormal hyperactive osteoclasts
Key Enzymes and Markers
- Cathepsin K = major collagenase
- TRAP (tartrate-resistant acid phosphatase) = serum marker
- Carbonic anhydrase II = generates H+ from CO2
- ClC-7 = chloride channel for electroneutrality
Evidence Base and Key Studies
Bone Resorption by Osteoclasts
- Landmark synthesis defining the osteoclast as a specialized macrophage polykaryon
- Established M-CSF, RANK ligand and osteoprotegerin as the principal differentiation regulators
- Described integrin-mediated cytoskeletal polarization creating the isolated resorption microenvironment
- Used osteopetrotic mutants to map genes controlling differentiation and resorptive capacity
Osteoclast Differentiation and Activation
- Authoritative review consolidating the RANK signalling pathway in osteoclasts
- Confirmed osteoclasts arise from the monocyte/macrophage haematopoietic lineage
- Positioned OPG as the soluble decoy receptor that neutralises RANK ligand
- Mapped how hormonal signals converge on RANKL to control bone mass
OPG Ligand (RANKL) Is the Osteoclast Differentiation Factor
- Discovery paper identifying OPG ligand (RANKL) as a TNF-family cytokine
- RANKL replaced the requirement for stromal cells, vitamin D3 and glucocorticoids in osteoclastogenesis co-culture
- Directly activated mature osteoclasts and produced hypercalcaemia when given to mice
- OPG blocked all RANKL effects in vitro and in vivo, defining the activator-decoy pair
Denosumab for Prevention of Fractures (FREEDOM Trial)
- Randomised placebo-controlled trial of 7868 postmenopausal women, T-score -2.5 to -4.0
- New vertebral fracture 2.3% vs 7.2% (RR 0.32) — a 68% relative reduction
- Hip fracture 0.7% vs 1.2% (HR 0.60); nonvertebral fracture 6.5% vs 8.0% (HR 0.80)
- 60 mg subcutaneous every 6 months; no osteonecrosis of the jaw and no excess infection or cancer over 36 months
Alendronate and Fracture Risk (Fracture Intervention Trial, FIT)
- Randomised trial of 2027 postmenopausal women with a prevalent vertebral fracture
- New morphometric vertebral fracture 8.0% vs 15.0% (RR 0.53) over 36 months
- Hip fracture RR 0.49 and wrist fracture RR 0.52 versus placebo
- No excess upper-gastrointestinal adverse events versus placebo
Vertebral Fractures After Discontinuation of Denosumab
- Post hoc analysis of 1001 FREEDOM/Extension participants who stopped denosumab
- Vertebral fracture rate rose from 1.2 to 7.1 per 100 participant-years after discontinuation
- Of those fracturing off-treatment, 60.7% had multiple vertebral fractures vs 38.7% after placebo
- Prior vertebral fracture raised the odds of multiple fractures 3.9-fold