Programmed Cell Death - Pathways and Relevance
- APOPTOSIS is PROGRAMMED cell death - an ACTIVE, energy-dependent, tightly REGULATED process ('cell suicide') - distinct from NECROSIS, which is passive, unregulated death from injury. Morphologically apoptosis causes cell SHRINKAGE, chromatin condensation, DNA fragmentation, membrane BLEBBING and formation of APOPTOTIC BODIES that are phagocytosed WITHOUT provoking inflammation, whereas necrosis causes cell swelling, membrane rupture and INFLAMMATION.
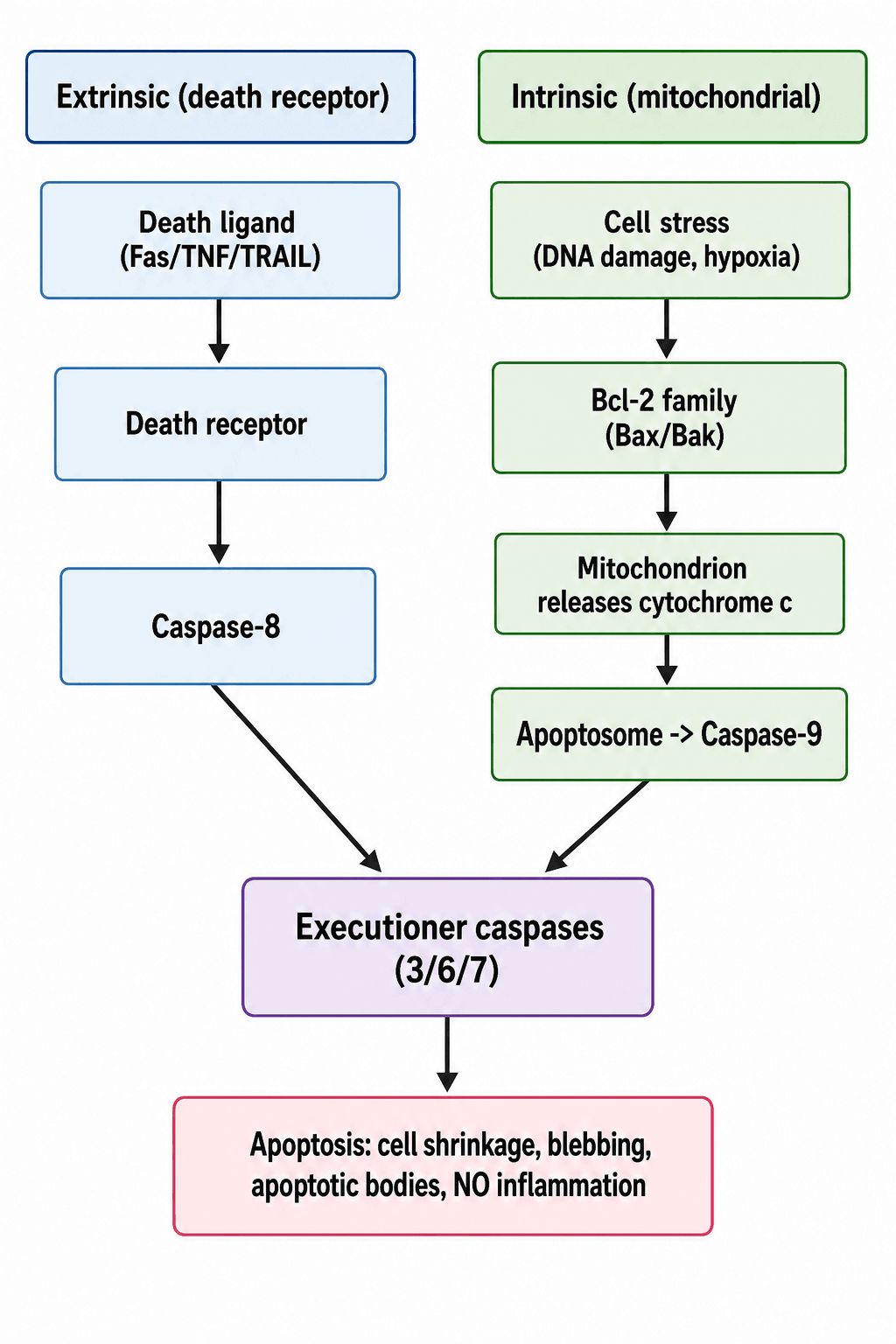
- Apoptosis proceeds via two pathways that converge on EXECUTIONER CASPASES (caspase-3/6/7), which dismantle the cell. The INTRINSIC (MITOCHONDRIAL) pathway is triggered by internal stress (DNA damage, hypoxia, oxidative/ER stress, growth-factor withdrawal), is regulated by the BCL-2 FAMILY (pro-apoptotic Bax/Bak versus anti-apoptotic Bcl-2), and proceeds via mitochondrial release of CYTOCHROME C, formation of the APOPTOSOME (with Apaf-1) and activation of CASPASE-9.
- The EXTRINSIC (DEATH-RECEPTOR) pathway is triggered when death ligands (FAS ligand, TNF, TRAIL) bind their DEATH RECEPTORS on the cell surface, forming a death-inducing signalling complex that activates CASPASE-8, which then activates the executioner caspases.
- Apoptosis is essential in NORMAL musculoskeletal biology: in the GROWTH PLATE, apoptosis of HYPERTROPHIC CHONDROCYTES is a necessary step in ENDOCHONDRAL OSSIFICATION (the cells die and are replaced by bone); in LIMB DEVELOPMENT, INTERDIGITAL apoptosis separates the digits (its failure contributes to SYNDACTYLY); and apoptosis participates in normal bone remodelling (osteoblast/osteocyte turnover).
- DYSREGULATED apoptosis features in orthopaedic DISEASE: excessive CHONDROCYTE APOPTOSIS contributes to cartilage loss in OSTEOARTHRITIS; OSTEOCYTE APOPTOSIS is central to OSTEONECROSIS/avascular necrosis and to glucocorticoid-induced and disuse bone loss; and apoptosis is involved in tendon degeneration, intervertebral disc degeneration and fracture healing.
- Apoptosis is also a THERAPEUTIC lever: chemotherapy and radiotherapy kill tumour cells largely by inducing apoptosis (and tumours resist by evading it, e.g. p53 loss or Bcl-2 overexpression), and bisphosphonates act partly by PROMOTING OSTEOCLAST apoptosis (while reducing osteocyte/osteoblast apoptosis) - so understanding apoptosis underlies several orthopaedic-relevant treatments.
- “Apoptosis = programmed, regulated, NO inflammation (shrinkage, blebbing, apoptotic bodies); necrosis = passive, inflammation (swelling, rupture).
- “Intrinsic (mitochondrial): Bcl-2 family -> cytochrome c -> apoptosome -> caspase-9. Extrinsic (death receptor): Fas/TNF/TRAIL -> caspase-8. Both -> executioner caspases (3/6/7).
- “Orthopaedic roles: growth-plate hypertrophic chondrocyte apoptosis (endochondral ossification), interdigital apoptosis (digit separation), chondrocyte apoptosis in OA, osteocyte apoptosis in osteonecrosis.
Active, regulated; cell shrinkage, blebbing, apoptotic bodies; NO inflammation; usually single cells.
Passive, unregulated injury; cell swelling, membrane rupture, spilled contents -> inflammation; usually groups of cells.
Mechanisms: Intrinsic & Extrinsic Pathways
Apoptosis is executed by caspases (cysteine-aspartate proteases). Two upstream pathways converge on the executioner caspases (3, 6, 7) that dismantle the cell:
- Intrinsic (mitochondrial) pathway: internal stress - DNA damage, hypoxia, oxidative/ER stress, growth-factor withdrawal - shifts the balance of the BCL-2 family toward pro-apoptotic Bax/Bak, which permeabilise the mitochondrial outer membrane to release cytochrome c; this assembles the apoptosome (with Apaf-1) and activates caspase-9. (p53 is a key trigger.)
- Extrinsic (death-receptor) pathway: death ligands - Fas ligand, TNF, TRAIL - bind their cell-surface death receptors, forming a death-inducing signalling complex that activates caspase-8. Both caspase-8 and caspase-9 then activate the executioner caspases, producing the characteristic shrinkage, blebbing and apoptotic bodies.
Why Apoptosis Causes No Inflammation: Phosphatidylserine and Efferocytosis
The defining feature the topic repeats - apoptotic bodies are "phagocytosed without inflammation" - has a mechanism the examiner wants. The dying cell keeps its membrane intact and flips phosphatidylserine from the inner to the outer leaflet, displaying it as an "eat me" signal.
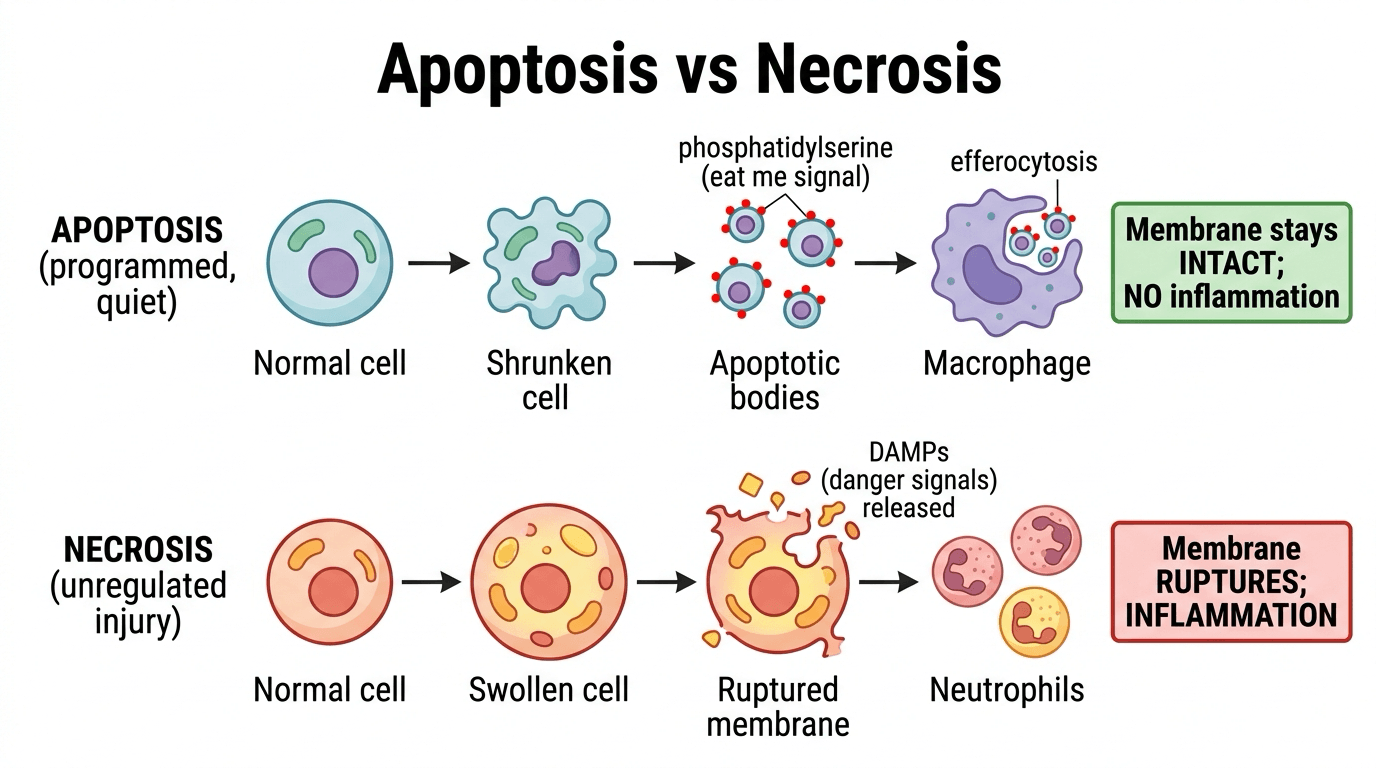
Macrophages and neighbouring cells recognise the externalised phosphatidylserine and rapidly engulf the apoptotic cell and its bodies (efferocytosis) before the membrane ruptures, so the pro-inflammatory intracellular contents are never released. Efferocytosis is itself anti-inflammatory (it releases TGF-beta and IL-10).
In necrosis the membrane ruptures and DAMPs (danger signals) spill out, provoking inflammation. This is exactly why apoptotic chondrocyte or osteocyte death is "quiet", whereas necrosis incites an inflammatory and reparative response.
Phosphatidylserine externalisation is both the "eat me" flag detected by annexin V in the lab and the trigger for non-inflammatory efferocytosis in vivo - the single molecular reason apoptosis, unlike necrosis, does not cause inflammation.
Apoptosis in Musculoskeletal Biology & Disease
- Growth plate / endochondral ossification: apoptosis of hypertrophic chondrocytes in the hypertrophic zone is an essential step - the cells die and are replaced by invading bone, allowing longitudinal growth.
- Limb development: interdigital apoptosis removes the webbing between digits to separate the fingers and toes; failure of this programmed cell death contributes to syndactyly.
- Bone remodelling: regulated apoptosis of osteoblasts and osteocytes is part of normal bone turnover.
- Osteoarthritis: excessive chondrocyte apoptosis contributes to cartilage matrix loss and joint degeneration.
- Osteonecrosis / avascular necrosis: osteocyte apoptosis (from ischaemia, steroids, alcohol) is central to the pathogenesis; glucocorticoids and disuse also drive osteocyte/osteoblast apoptosis and bone loss.
- Other: apoptosis is implicated in tendon degeneration, intervertebral disc degeneration and aspects of fracture healing.
- Therapeutic relevance: chemotherapy/radiotherapy kill tumour cells largely by inducing apoptosis (tumours evade it via p53 loss or Bcl-2 overexpression); bisphosphonates promote osteoclast apoptosis (reducing resorption) while reducing osteocyte/osteoblast apoptosis.
Demonstrating Apoptosis: The Laboratory Assays
Recognising the morphology of apoptosis is only half the basic-science question. You also need to know how apoptosis is demonstrated, because distinguishing apoptosis from necrosis in cartilage, bone or a research model depends on these assays.
- DNA fragmentation -> TUNEL assay (labels the cut DNA ends in situ) and DNA "laddering" on gel electrophoresis (internucleosomal cleavage gives regular fragments in multiples of about 180 base pairs).
- Phosphatidylserine externalisation -> annexin V binding (flow cytometry), paired with a viability dye to separate early apoptosis from necrosis.
- Active executioner caspase -> cleaved caspase-3 immunostaining.
- Morphology -> electron microscopy (condensed chromatin, apoptotic bodies) and light microscopy (pyknosis, karyorrhexis).
These assays let you quantify apoptosis and separate it from necrosis - for example counting apoptotic chondrocytes in osteoarthritic cartilage or apoptotic osteocytes in osteonecrotic or glucocorticoid-treated bone - which is how the disease associations in this topic were established and how candidate therapies are tested.
Mnemonics & Memory Aids
APOPTOSIS
Hook:APOPTOSIS = Active, Programmed, Organised, No inflammation.
8 vs 9
Hook:Caspase-8 = extrinsic (outside); caspase-9 = intrinsic (inside).
Clinical Decision Scenarios
Practise clinical reasoning and management decisions out loud
“What is the difference between apoptosis and necrosis, and what are the two apoptosis pathways?”
“Where is apoptosis important in orthopaedics, in both normal biology and disease?”
Apoptosis vs necrosis
- Apoptosis: active, regulated, no inflammation; shrinkage/blebbing/apoptotic bodies; single cells
- Necrosis: passive, injury, inflammation; swelling/rupture; groups of cells
Pathways
- Intrinsic (mitochondrial): stress -> Bcl-2 family (Bax/Bak) -> cytochrome c -> apoptosome -> caspase-9
- Extrinsic (death receptor): Fas/TNF/TRAIL -> caspase-8
- Both -> executioner caspases (3/6/7)
Normal orthopaedic roles
- Growth plate: hypertrophic chondrocyte apoptosis (endochondral ossification)
- Limb development: interdigital apoptosis (digit separation; failure -> syndactyly)
- Bone remodelling (osteoblast/osteocyte turnover)
Disease & therapy
- OA: chondrocyte apoptosis; osteonecrosis & steroid/disuse loss: osteocyte apoptosis
- Tendon/disc degeneration; fracture healing
- Chemo/radiotherapy induce apoptosis; bisphosphonates promote osteoclast apoptosis
Evidence & Key Studies
Microbiota metabolite lithocholic acid in cancer: apoptosis via intrinsic and extrinsic, caspase-dependent pathways (review)
- Apoptosis is induced through both intrinsic and extrinsic pathways and is (at least partly) caspase-dependent.
- Intrinsic apoptosis involves mitochondrial dysfunction and endoplasmic-reticulum stress, with the cell's stress state determining the response.
- In cancer cells the microbiota metabolite lithocholic acid induces selective apoptosis (sparing normal cells) via these intrinsic/extrinsic caspase pathways, illustrating the conserved apoptosis machinery.
IGFBP3 in osteoarthritis: modulation of chondrocyte proliferation and apoptosis
- Chondrocyte APOPTOSIS (alongside proliferation and signalling) is a key process in osteoarthritis cartilage homeostasis and degeneration.
- IGFBP3 modulates chondrocyte apoptosis through IGF-1-dependent and -independent mechanisms, with both protective and disease-promoting actions reported.
- Illustrates the orthopaedic relevance of regulated apoptosis in joint degeneration and as a potential therapeutic target.
The intrinsic/extrinsic, caspase-dependent mechanism of apoptosis (with mitochondrial and ER-stress involvement) comes from the cited Darmadi review, and the role of chondrocyte apoptosis in osteoarthritis from the cited Chen review. The apoptosis-versus-necrosis distinction, the Bcl-2/cytochrome c/caspase machinery, and the roles of apoptosis in the growth plate, limb development and osteonecrosis are standard, well-established cell-biology teaching. (See also our Bone Healing, Physis/Growth Plate, Osteonecrosis and Osteoarthritis topics.)