Progressive Neurodevelopmental Regression
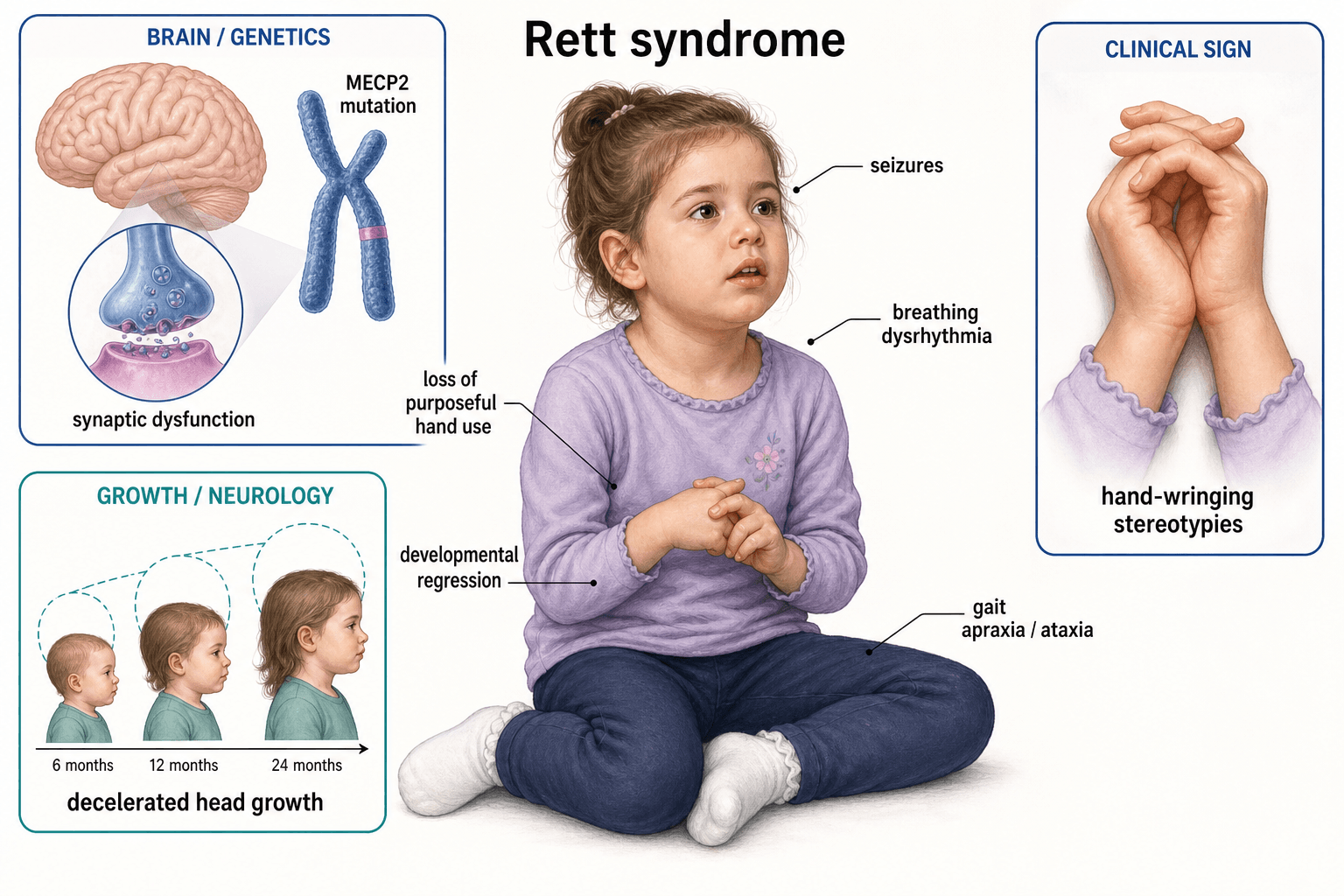
- MECP2 Gene: X-linked. Males usually not viable.
- Regression: Normal development then regression at 6-18 months.
- Hand Stereotypies: Classic hand-wringing.
- Scoliosis: Nearly universal, often severe.
- Non-ambulatory: Most patients are wheelchair-bound.
- “MECP2 mutation
- “Hand-wringing stereotypies
- “Scoliosis is nearly universal
- “Females almost exclusively
Scoliosis surgery in Rett Syndrome has unique considerations.
- Patients are non-communicative - pain assessment is difficult.
- High complication rate.
- Respiratory compromise common.
- Long fusions (often to pelvis) are needed.
- QOL benefits for sitting balance and caregiving.
- Age
- 6-18 months
- Features
- Decelerated development, hypotonia
- Age
- 1-4 years
- Features
- Loss of skills, hand stereotypies, seizures
- Age
- Preschool-adult
- Features
- Stable, seizures, scoliosis develops
- Age
- Variable
- Features
- Reduced mobility, severe scoliosis
SHFOrthopaedic Issues
Hook:SHF - Spine, Hips, Feet.
CRSSurgery Challenges
Hook:CRS - Communication, Respiratory, Seizures.
Overview/Epidemiology
Rett Syndrome is a neurodevelopmental disorder with regression.
- Genetics: X-linked dominant. Mutations in MECP2 gene (Xq28).
- Incidence: 1 in 10,000-15,000 females.
- Sex: Almost exclusively females. Males with MECP2 mutations usually die in utero.
- Natural History: Normal development followed by regression at 6-18 months.
Pathophysiology
Why Scoliosis Develops
- Central hypotonia and poor motor control.
- Asymmetric posture and muscle activity.
- Curves are typically long C-shaped thoracolumbar.
- Pelvic obliquity is common.
- Progression is relentless in most.
Hip Dysplasia
- Hypotonia + abnormal posture → subluxation.
- Often asymptomatic.
MeCP2 Function & Why Severity Varies
The topic states the cause is MECP2 and that the protein is "involved in gene regulation" without explaining what MeCP2 does or why the phenotype varies so widely - both examinable.
- What MeCP2 does. MeCP2 (methyl-CpG-binding protein 2) binds methylated CpG dinucleotides and acts as a transcriptional regulator (classically a repressor recruiting histone deacetylase / Sin3A, but it both represses and activates genes). It is most abundant in mature neurons, where it is needed for synaptic maturation and maintenance - not for the initial wiring of the brain.
- Why regression follows a normal period. Because MeCP2 is required for maintaining mature synaptic function rather than building the early nervous system, the infant develops normally for the first 6-18 months, then regresses as the maturing brain fails without functional MeCP2 - the hallmark "normal then lost" course.
- Why females survive and severity varies (X-inactivation). Females are mosaics: each cell randomly inactivates one X, so a female with a heterozygous MECP2 mutation has a mixture of cells expressing the normal or the mutant allele. The degree and pattern of X-inactivation skewing - together with the specific mutation - largely explains the wide phenotypic variability between affected girls.
- Why males are usually not viable, and the exceptions. A hemizygous male has the mutation in every cell (no normal-allele mosaic), so a classic MECP2 mutation is usually lethal or causes severe neonatal encephalopathy. Rare male survivors occur with Klinefelter (47,XXY), somatic mosaicism, or milder mutations.
This molecular picture underpins both the diagnosis (regression-then-stabilisation) and the move toward disease-modifying therapy.
MeCP2 binds methylated CpG and regulates transcription (recruiting HDAC/Sin3A); it is needed to maintain mature neurons, so the brain develops normally then regresses. X-inactivation mosaicism (skewing) plus the specific mutation explain the wide variability between girls; a hemizygous male has no normal-allele mosaic so it is usually lethal (survivors: Klinefelter/47,XXY, somatic mosaicism, or milder mutations).
Classification Systems
Clinical Stages (Hagberg)
- Stage I (Early Onset Stagnation): 6-18 months. Developmental deceleration.
- Stage II (Rapid Developmental Regression): 1-4 years. Loss of hand skills, speech. Hand stereotypies begin.
- Stage III (Pseudostationary): Preschool-adult. Stable phase. Seizures, breathing irregularities. Scoliosis develops.
- Stage IV (Late Motor Deterioration): Reduced mobility, severe scoliosis, muscle wasting.
Clinical Assessment
- Age of regression.
- Current mobility and communication status.
- Seizure history.
- Respiratory status.
- Feeding issues.
- General: Non-communicative. May be agitated.
- Hand Stereotypies: Hand-wringing, hand-mouthing.
- Spine: Scoliosis, often severe.
- Hips: Assess for subluxation.
- Feet: Equinovarus.
- Respiratory: Breathing irregularities (hyperventilation, breath-holding).
Investigations
- MECP2 mutation: Confirmatory.
- Spine X-ray: Scoliosis assessment.
- Hip X-ray: Subluxation.
- ECG: QT prolongation can occur.
- Sleep study if needed.
Differential Diagnosis
- Genetics
- MECP2 (Xq28), X-linked dominant
- Discriminating features
- Regression after normal development, hand-wringing stereotypies, acquired microcephaly, near-universal scoliosis, almost exclusively female
- Genetics
- UBE3A (15q11-q13)
- Discriminating features
- Happy/excitable demeanour, ataxic wide-based gait, severe speech deficit; no true regression, no hand-wringing
- Genetics
- CDKL5 (Xp22)
- Discriminating features
- Early-onset epilepsy in first months (before regression typical of RTT); previously called early-seizure RTT variant
- Genetics
- FOXG1 (14q12)
- Discriminating features
- Congenital onset (no normal period), early microcephaly, dyskinesia; previously called congenital RTT variant
- Genetics
- Heterogeneous / polygenic
- Discriminating features
- Social-communication deficits without the discrete regression-then-stabilisation course or stereotypic hand-wringing
- Genetics
- Non-genetic (acquired)
- Discriminating features
- Static, non-progressive motor disorder; no regression, no hand stereotypies; spasticity pattern reflects timing of injury
MECP2 mutation is confirmatory in most classic cases; classic hand-wringing/hand-mouthing stereotypies; regression after a period of normal development; almost exclusively female.
male patient (very rare in classic RTT); absent regression history; absent hand stereotypies; congenital onset with no normal period (consider FOXG1); seizures dominating from the first months of life (consider CDKL5).
Management Algorithm
Scoliosis Management
- Observation: Mild curves.
- Bracing (Seating Support): Does not prevent progression but aids sitting.
- Surgery: Posterior spinal fusion for curves greater than 40-50 degrees. Usually T2-pelvis.
Key Surgical Considerations
- ECG for QT prolongation - affects anesthetic drug choice.
- Seizure control on current medications.
- Nutritional status - many have gastrostomy.
- Respiratory function - breathing irregularities common.
- Avoid QT-prolonging anesthetic drugs.
- Careful monitoring for arrhythmias.
- High blood loss expected with long fusions.
- Pain assessment in non-communicative patient.
- Respiratory monitoring in ICU.
- Early mobilization to seating.
- Careful wound care.
Surgical Techniques
Posterior Spinal Fusion
Indications: Progressive scoliosis greater than 40-50 degrees.
Technique: Posterior approach. Long fusion (T2 to pelvis). Pelvic fixation with iliac screws or S2-alar-iliac screws. Pedicle screw constructs.
Considerations: High complication rate. Respiratory issues common. Non-communicative patients - pain assessment difficult. ICU post-op.
Foot Deformity in Rett Syndrome
The mnemonics, examination and cheat sheet repeatedly list equinovarus feet as a core orthopaedic problem, but the topic never develops their assessment or management.
- The deformity. The commonest foot deformity is a neuromuscular equinovarus (and sometimes equinus or planovalgus), driven by spasticity/dystonia and muscle imbalance with a relentless tendency to become fixed and rigid in a largely non-ambulatory child; it interferes with footwear, foot positioning on a wheelchair footplate, transfers and skin care rather than with walking.
- Non-operative first. Stretching, ankle-foot orthoses (AFOs) and accommodative footwear maintain a plantigrade, braceable foot; botulinum toxin can reduce dynamic spastic/dystonic deforming force in a flexible foot and delay surgery.
- Surgery for the fixed, problematic foot. When the deformity is fixed and impairs shoeing/positioning or causes skin breakdown, options are soft-tissue releases and tendon lengthening/transfer for a still- correctable foot, and, for a rigid foot in the older child, a bony procedure such as a triple arthrodesis (or osteotomies) to obtain a stable, plantigrade, shoeable foot.
- The realistic goal. In a non-ambulator the aim is a painless, plantigrade, braceable foot for sitting, transfers and skin care - not a normal gait - so intervention is driven by function and skin, not the radiograph. (Operative correction of the spastic equinovarus foot is detailed in our Spastic Equinovarus Foot topic.)
Equinovarus in Rett is a neuromuscular, often progressive-to-fixed deformity in a non-ambulator - the goal is a painless, plantigrade, braceable foot for footwear/positioning/skin care, not gait. Start with stretching/AFO ± botulinum toxin; for a fixed foot use soft-tissue release/tendon transfer, escalating to triple arthrodesis for the rigid foot. Decide on function and skin, not the radiograph.
RETTRett Features
Hook:RETT - Regression, MECP2, Stereotypies, Truncal (scoliosis).
Complications
- Context
- Perioperative, disease-related
- Management
- BiPAP, careful anesthesia
- Context
- Wound, UTI
- Management
- Prophylaxis, antibiotics
- Context
- Long fusions
- Management
- Revision if symptomatic
- Context
- Anesthesia risk
- Management
- ECG, avoid QT-prolonging drugs
Postoperative Care
- ICU Monitoring: Respiratory.
- Pain Assessment: Difficult. Use behavioral scales.
- Mobilization: Sitting as tolerated.
- Long-Term: Orthotic support, ongoing medical care.
Outcomes/Prognosis
- Life Expectancy: Variable. Many survive to 40s-50s.
- Scoliosis Surgery: Improves sitting, caregiving ease. May not improve survival.
- QOL: Difficult to assess. Benefits often for caregivers.
Guidelines, Registries & Global Practice
Global epidemiology: Classic Rett syndrome affects approximately 1 in 10,000 to 15,000 live female births worldwide, making it one of the commonest genetic causes of severe intellectual disability in girls. Distribution is global with no major ethnic predilection.
Side-by-side guidance:
- Focus
- Diagnosis
- Key recommendation
- Clinical criteria define classic vs atypical RTT; MECP2 testing confirms but is not required for diagnosis
- Focus
- Neuromuscular scoliosis
- Key recommendation
- Long instrumented posterior fusion to the pelvis for progressive curves; expert consensus on perioperative optimisation
- Focus
- Paediatric spinal deformity
- Key recommendation
- Surveillance-based pathway; surgery centralised to specialist paediatric spine units
- Focus
- Operative technique
- Key recommendation
- Pedicle-screw constructs and pelvic fixation (iliac or S2-alar-iliac) as the standard for neuromuscular curves
Population-based registries (notably long-running Rett databases) have provided the strongest outcome data, including the survival benefit of fusion for severe early-onset scoliosis (Downs et al. 2015). National spine and neuromuscular registries inform implant and complication benchmarking.
In well-resourced systems, care is multidisciplinary (neurology, orthopaedics, respiratory, genetics, rehabilitation) with early genetic confirmation, structured hip/spine surveillance, and fusion at high-volume paediatric spine centres with ICU support. In limited-resource settings genetic testing may be unavailable (diagnosis remains clinical), surveillance is opportunistic, and access to safe long-construct surgery with intensive perioperative care is restricted - shifting management toward seating/postural support and supportive care.
Controversies & Areas of Uncertainty
- Timing and extent of spinal fusion. Registry data (Downs et al. 2015) support fusion of severe early-onset curves for a survival benefit, but the optimal age and Cobb threshold remain debated. Conventional teaching fuses progressive curves over 40 to 50 degrees, yet some advocate earlier intervention in rapidly progressing skeletally immature girls.
- Fusionless vs definitive fusion in the immature spine. Growth-friendly constructs (traditional growing rods, magnetically controlled rods) and minimally invasive bipolar techniques can defer arthrodesis while preserving trunk/thoracic growth, but carry repeated-procedure burden and their own complication profile; the optimal strategy is unsettled.
- Role of bracing. Bracing does not alter the natural history of the curve and is used only for seating/postural support, not curve control - a frequent exam trap.
- Hip surveillance vs intervention. Hip displacement is common, but most hips are pain-free even when subluxated. There is no consensus on a migration-percentage threshold mandating surgery in non-ambulators; management is largely symptom-driven.
- Disease-modifying therapy. Trofinetide (an IGF-1 analogue) is now approved in some jurisdictions for the neurological phenotype; it does not address established orthopaedic deformity, and its long-term impact on scoliosis progression is unknown.
MCQ Practice Points
Q: What gene is mutated in Rett Syndrome? A: MECP2 gene on Xq28.
Q: Why is Rett Syndrome almost exclusively seen in females? A: It is X-linked dominant. Males with MECP2 mutations usually have embryonic lethality.
Q: What is the incidence of scoliosis in Rett Syndrome? A: 80-90%.
Q: What is the classic hand movement in Rett Syndrome? A: Hand-wringing stereotypies.
Q: What is the extent of fusion for scoliosis in Rett Syndrome? A: T2 to pelvis with pelvic fixation (S2-alar-iliac or iliac screws).
Q: What cardiac issue should be checked before scoliosis surgery in Rett? A: QT prolongation on ECG - affects anesthetic drug choice.
Self-Assessment Quiz
Additional Quiz Questions
Viva Scenarios
Practise clinical reasoning and management decisions out loud
“10-year-old female with Rett Syndrome. Thoracolumbar scoliosis of 65 degrees with pelvic obliquity. Non-ambulatory, non-communicative. Seizures controlled on medication.”
“What is the genetic cause of Rett Syndrome?”
“Same patient has bilateral hip subluxation (MP 40%). Should this be treated surgically?”
GENETICS
- MECP2 Gene
- Xq28
- X-linked Dominant
- Females almost exclusively
CLINICAL
- Regression 6-18mo
- Hand stereotypies
- Seizures
- Non-communicative
ORTHOPAEDIC
- Scoliosis 80-90%
- Hip subluxation
- Equinovarus feet
- Pelvic obliquity
SCOLIOSIS SURGERY
- T2-pelvis fusion
- Pelvic fixation essential
- High complication rate
- ICU post-op care
PRE-OP CHECKS
- ECG for QT prolongation
- Seizure control
- Respiratory status
- Nutrition assessment
PROGNOSIS
- Survival to 40s-50s
- Surgery improves sitting
- Non-ambulatory most
- QOL benefits caregivers
Evidence Base
- Identified de novo mutations in X-linked MECP2 (Xq28) as the cause of Rett syndrome
- First disease-causing mutations reported, in the methyl-binding and transcription-repression domains
- Confirmed X-linked dominant mechanism with abnormal epigenetic regulation
- International consensus revising the 2002 diagnostic criteria for classic and atypical RTT
- Reinforces that RTT remains a clinical diagnosis, independent of molecular findings
- Defines required main criteria: regression then recovery/stabilisation, loss of hand skills, loss of language, gait abnormalities, hand stereotypies
- Survey of 258 families; scoliosis present in 119 patients
- Incidence rises with age, most commonly during the second decade
- Bracing was largely unsuccessful at controlling curve progression in adolescents
- Long C-shaped thoracolumbar neuromuscular curve; reported incidence 36 to 100 percent
- Onset usually before age 8; rapid progression early in the second decade
- Surgical indication is curve progression beyond a 40 to 45 degree Cobb angle, or pain/loss of function
- Clinic cohort of 31 females: 48 percent had hip migration percentage of 30 percent or more
- 27 of 31 had scoliosis and 20 had a Cobb angle over 30 degrees
- Recommends early, repeated radiological surveillance of hips and spine in all young patients
- Prospective long-term follow-up of 23 girls after spinal fusion (mean 74 months)
- Improved sitting balance, weight distribution, fewer seating supports and reduced rest time
- Parents reported better seating, daily activities and cosmesis
- Population-based cohort of 140 females with severe scoliosis (Cobb 45 degrees or more before adulthood)
- Spinal fusion associated with lower mortality (adjusted HR 0.30, 95 percent CI 0.12 to 0.74)
- Survival benefit greatest for early-onset scoliosis (HR 0.17, 95 percent CI 0.06 to 0.52)
- Neuromuscular scoliosis carries the highest complication rate of all scoliosis (6 to 75 percent)
- Pulmonary complications dominate (up to 23 to 29 percent); implant-related next (13 to 23 percent)
- Specifically flags conduction abnormalities in Rett syndrome as a potentially lethal, screenable risk