Defective Globin Chain Synthesis with Skeletal Manifestations
Thalassemia Classification
Critical Must-Knows
- Beta Thalassemia Major: Most severe form, transfusion-dependent from early childhood.
- Marrow Expansion: Causes widened medullary cavities, cortical thinning, pathological fractures.
- Osteoporosis: Multifactorial - marrow expansion, iron toxicity, hypogonadism, deferoxamine.
- Hair-on-end Skull: Classic radiographic finding from diploic expansion.
- Extramedullary Hematopoiesis: Can cause spinal cord compression.
Clinical Pearls
- "Hair-on-end skull - occipital spared (no marrow)
- "Chipmunk facies from maxillary expansion
- "Osteoporosis common even in young patients
- "DEXA screening and bisphosphonates for bone health
- "EMH can compress spinal cord - surgical emergency
Osteoporosis in Thalassemia
Osteoporosis is a major cause of morbidity, even in young patients.
- Causes: Marrow expansion, iron toxicity, deferoxamine, hypogonadism, vitamin D deficiency
- Prevalence: 40-80% of adult patients have osteoporosis
- Complications: Vertebral compression fractures (10-20%), long bone fractures
- Management: DEXA screening, bisphosphonates, vitamin D/calcium, hormone replacement
Inheritance
Geography
Osteoporosis Prevalence
Marrow Expansion
At a Glance
Thalassemia is an inherited autosomal recessive hemoglobinopathy with defective alpha or beta globin chain synthesis, most prevalent in the Mediterranean belt, Middle East, and Southeast Asia. Beta thalassemia major is the most severe form, requiring lifelong transfusions from early childhood. Orthopaedic manifestations result from marrow expansion (6-fold increase in erythroid precursors): hair-on-end skull (occipital spared as it lacks marrow), chipmunk facies (maxillary expansion), widened medullary cavities with cortical thinning, and pathological fractures. Osteoporosis affects 40-80% of adult patients due to multiple factors—marrow expansion, iron toxicity, deferoxamine therapy, and hypogonadism—causing vertebral compression fractures in 10-20%. Extramedullary hematopoiesis can cause spinal cord compression requiring emergency treatment.
MOFHThalassemia Bone Changes - MOFH
| M | Marrow Expansion Widened medullary cavities, thin cortices |
| O | Osteoporosis 40-80% of adults affected |
| F | Facial Changes Chipmunk facies, frontal bossing |
| H | Hair-on-end Classic skull X-ray appearance |
| M | Marrow Expansion Widened medullary cavities, thin cortices | F | Facial Changes Chipmunk facies, frontal bossing |
| O | Osteoporosis 40-80% of adults affected | H | Hair-on-end Classic skull X-ray appearance |
Hook:MOFH - Marrow, Osteoporosis, Facial, Hair-on-end
MIDHECauses of Osteoporosis in Thalassemia - MIDHE
| M | Marrow expansion Mechanical disruption of bone formation |
| I | Iron overload Toxic to osteoblasts |
| D | Deferoxamine Chelation therapy affects bone |
| H | Hypogonadism Iron deposition in pituitary/gonads |
| E | Endocrine Vitamin D deficiency, thyroid dysfunction |
| M | Marrow expansion Mechanical disruption of bone formation | H | Hypogonadism Iron deposition in pituitary/gonads |
| I | Iron overload Toxic to osteoblasts | E | Endocrine Vitamin D deficiency, thyroid dysfunction |
| D | Deferoxamine Chelation therapy affects bone |
Hook:MIDHE causes weak bones in thalassemia
THALASSEMIAThalassemia vs Sickle Cell - THALASSEMIA
| T | Transfusion-dependent Beta major needs regular transfusions |
| H | Hair-on-end Both can show this (thalassemia more common) |
| A | AVN less common Unlike sickle cell where AVN is very common |
| L | Lacks vaso-occlusion No acute pain crises like sickle cell |
| T | Transfusion-dependent Beta major needs regular transfusions | A | AVN less common Unlike sickle cell where AVN is very common |
| H | Hair-on-end Both can show this (thalassemia more common) | L | Lacks vaso-occlusion No acute pain crises like sickle cell |
Hook:THAL - No vaso-occlusion differentiates from sickle cell
Overview/Epidemiology
Thalassemia is a group of inherited hemoglobin disorders characterized by reduced or absent synthesis of one or more globin chains.
Genetics:
- Autosomal recessive inheritance
- Alpha thalassemia: Deletion of 1-4 alpha globin genes (chromosome 16)
- Beta thalassemia: Point mutations in beta globin gene (chromosome 11)
Epidemiology:
- Most common inherited hemoglobin disorder worldwide
- Highest prevalence: Mediterranean, Middle East, Southeast Asia, Indian subcontinent, Africa
- Carrier frequency: Up to 30% in endemic areas
- Annual affected births: 60,000-70,000 globally
Classification by Severity:
| Type | Genotype | Clinical Features |
|---|---|---|
| Beta Major (Cooley's) | β0/β0 or β0/β+ | Severe anemia, transfusion-dependent |
| Beta Intermedia | Variable | Moderate anemia, variable transfusion needs |
| Beta Minor (Trait) | β/β0 or β/β+ | Mild microcytic anemia, carrier state |
| Alpha Thalassemia | 1-4 gene deletions | Variable: silent carrier to hydrops fetalis |
Pathophysiology
Hematological Pathophysiology:
Ineffective Erythropoiesis
The fundamental defect in thalassemia is imbalanced globin chain production:
- Beta thalassemia: Reduced or absent beta chains → excess alpha chains
- Unpaired alpha chains precipitate → damage RBC membrane
- Premature destruction of RBC precursors in bone marrow
- Chronic hemolytic anemia triggers compensatory marrow expansion
- Erythroid precursors increase up to 6-fold
- Medullary cavity expansion and cortical thinning result
This process drives the skeletal manifestations unique to thalassemia.
Skeletal Pathophysiology:
The orthopaedic manifestations result from:
- Marrow Expansion: Widened medullary cavities, thin cortices, increased fragility
- Bone Resorption: Cancellous bone loss from osteoclast activity
- Iron Toxicity: Direct damage to osteoblasts and osteocytes
- Chelation Effects: Deferoxamine may impair bone metabolism
- Endocrine Dysfunction: Hypogonadism, hypothyroidism, diabetes from iron deposition
Classification
Beta Thalassemia Classification
By Genotype and Severity:
| Classification | Genotype | Clinical Phenotype |
|---|---|---|
| Major (Cooley's Anemia) | β0/β0, β0/β+, β+/β+ (severe) | Transfusion-dependent from infancy |
| Intermedia | Variable combinations | Moderate anemia, variable transfusions |
| Minor (Trait) | β/β0 or β/β+ | Mild microcytic anemia, asymptomatic |
Beta Major Features:
- Presents 6-12 months of age (after HbF decline)
- Severe anemia (Hb 3-6 g/dL untreated)
- Hepatosplenomegaly
- Growth retardation
- Skeletal changes from marrow expansion
Beta Intermedia Features:
- Variable severity
- May not require regular transfusions
- Still develop skeletal changes
- Iron overload from GI absorption (not transfusion)
Modern transfusion programs have reduced skeletal deformities.
Clinical Presentation
Skeletal Manifestations:
Craniofacial Changes
Skull:
- Hair-on-end appearance: Perpendicular spicules from diploe expansion
- Widened diploic space
- Thinned outer table
- Occipital sparing: No haematopoietic marrow in occipital bone
Face:
- Chipmunk facies: Maxillary hypertrophy
- Frontal bossing
- Prominent malar eminences
- Lateral orbital displacement
- Dental malocclusion
- Sinus obliteration (except ethmoid)
These changes are less common with modern transfusion protocols that suppress marrow expansion.
Systemic Manifestations Affecting Orthopaedics:
- Growth retardation: Short stature, delayed puberty
- Hypogonadism: Contributes to osteoporosis
- Splenomegaly: May need splenectomy (increases infection risk)
- Iron overload: Affects bone metabolism
Investigations
Laboratory Studies:
| Test | Expected Finding | Clinical Significance |
|---|---|---|
| CBC | Microcytic anemia (MCV low) | Severity indicates type |
| Hb electrophoresis | Increased HbA2, HbF | Diagnostic for beta thalassemia |
| Iron studies | High ferritin, high iron | Iron overload monitoring |
| LFTs | May be elevated | Hepatic iron deposition |
| Endocrine panel | Hypogonadism, hypothyroidism | Secondary complications |
| Vitamin D | Often deficient | Contributes to osteoporosis |
Imaging:
Plain Radiographs
Skull:
- Hair-on-end appearance
- Widened diploic space
- Occipital sparing
Spine:
- Osteopenia
- Striated vertebrae
- Compression fractures
- Biconcave deformities
Long Bones:
- Widened medullary cavities
- Cortical thinning
- Pathological fractures
X-ray remains useful for initial assessment and fracture detection.
Management
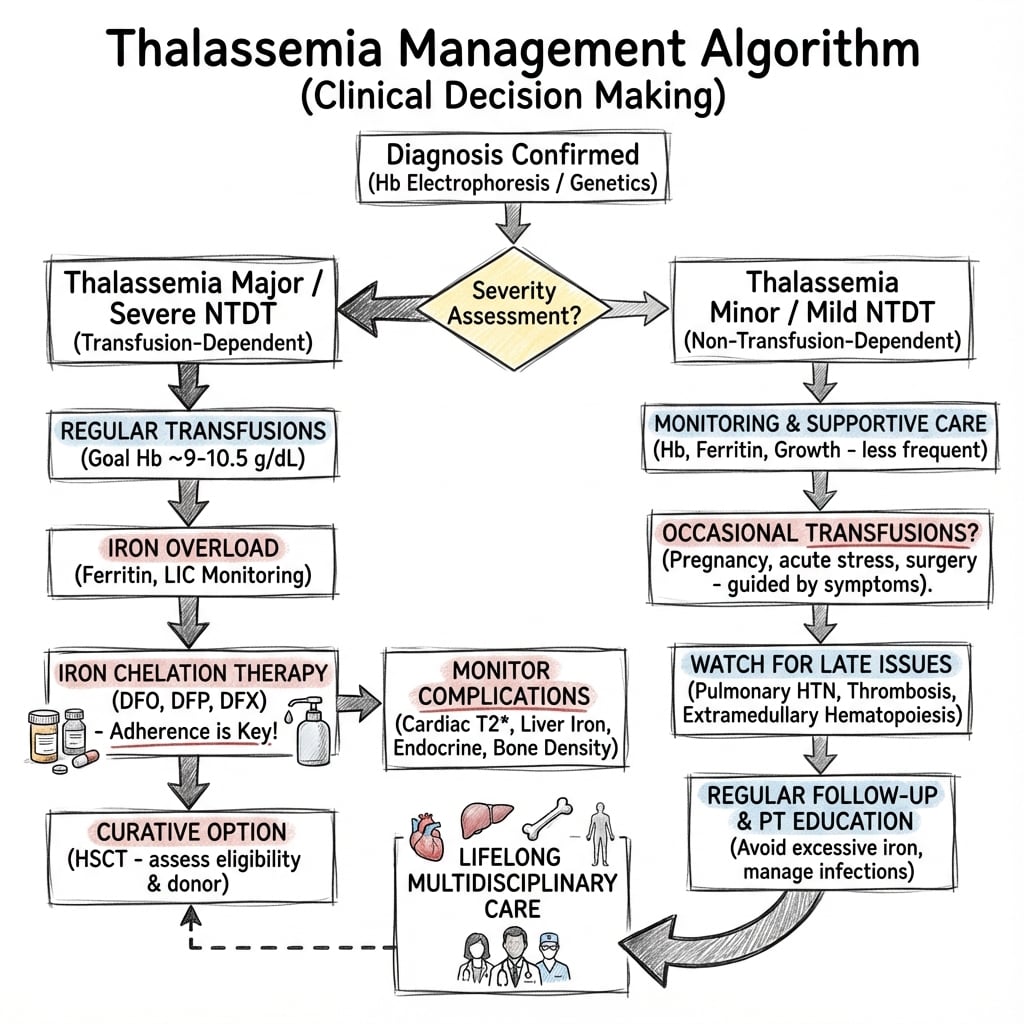
Perioperative Considerations:
- Coordinate with hematology for transfusion timing
- Check cardiac function (iron cardiomyopathy)
- Risk of venous thromboembolism (post-splenectomy especially)
- Immunocompromised if splenectomized
- Delayed wound and fracture healing possible
Medical Management
Transfusion Therapy:
- Beta major: Regular transfusions every 2-4 weeks
- Target: Pre-transfusion Hb of 9-10 g/dL
- Suppresses endogenous erythropoiesis
- Reduces marrow expansion and skeletal deformities
Iron Chelation:
- Deferoxamine (Desferal): SC or IV infusion, traditional agent
- Deferasirox (Exjade): Oral, once daily
- Deferiprone (Ferriprox): Oral, for cardiac iron
- Target: Ferritin less than 1000 μg/L
Bone Health:
- Calcium and vitamin D supplementation
- Bisphosphonates for osteoporosis (zoledronic acid, pamidronate)
- Hormone replacement for hypogonadism
- Regular DEXA monitoring
Curative Treatment:
- Bone marrow transplant (HLA-matched sibling donor)
- Gene therapy (emerging)
Modern comprehensive care has dramatically improved outcomes.
Surgical Considerations in Thalassemia
Preoperative Planning
Hematology Coordination:
- Preoperative transfusion to Hb 10-11 g/dL
- Assess cardiac function (iron cardiomyopathy)
- Check ferritin levels and chelation status
- Optimize coagulation (platelets, liver function)
Bone Quality Assessment:
- DEXA scan for bone density
- CT/MRI for bone architecture if significant surgery planned
- Anticipate poor bone quality for fixation
Preoperative Checklist
| Assessment | Target | Concern |
|---|---|---|
| Hemoglobin | 10-11 g/dL | Transfuse if low |
| Cardiac function | EF greater than 50% | Iron cardiomyopathy risk |
| Ferritin | Optimized chelation | Bleeding risk if high |
| Bone density | DEXA T-score | Implant choice affected |
Complications
Skeletal Complications
Osteoporosis:
- Most common orthopaedic complication
- Prevalence 40-80% in adults
- Affects quality of life significantly
Fractures:
- Vertebral compression fractures: 10-20%
- Long bone fractures: Increased risk
- Hip fractures: Major concern
- Delayed healing common
Deformity:
- Craniofacial changes (undertreated patients)
- Spinal deformity
- Short stature
Growth Disturbance:
- Height less than 3rd percentile common
- Delayed bone age
- Delayed puberty
Growth disturbance in thalassemia major reflects both marrow expansion diverting metabolic resources and endocrine dysfunction from iron overload.
Comparison: Thalassemia vs Sickle Cell Disease:
Thalassemia vs Sickle Cell Disease
| Feature | Thalassemia | Sickle Cell Disease |
|---|---|---|
| Globin chain quantity (reduced) | Globin chain quality (HbS) | |
| Marrow expansion, osteoporosis | Vaso-occlusion, infarction | |
| Uncommon | Very common (hip, shoulder) | |
| No | Yes (vaso-occlusive) | |
| Not increased | Increased (Salmonella) | |
| Classic finding | Can occur but less common |
Postoperative Care
Post-Surgery Management
Hematology Care:
- Continue transfusion protocol per hematology
- Resume chelation when safe (typically 24-48 hours post-op)
- Monitor hemoglobin and transfuse as needed
- DVT prophylaxis (increased thrombosis risk post-splenectomy)
Wound/Fracture Care:
- Expect delayed wound healing (poor vascularity)
- Monitor for infection (immunocompromised if splenectomized)
- Extended weight-bearing restrictions for fractures
- Consider bone stimulator for delayed union
Postoperative Monitoring
| Parameter | Target | Frequency |
|---|---|---|
| Hemoglobin | 9-10 g/dL | Daily initially |
| Wound healing | Monitor for infection | Regular inspection |
| Fracture healing | Serial X-rays | 6-8 weekly |
| DVT prophylaxis | Per protocol | Extended duration |
Outcomes
Orthopaedic Outcomes
Fracture Healing:
- Delayed union is common
- Nonunion rate higher than general population
- Good outcomes with appropriate fixation and protection
Osteoporosis Treatment:
- Bisphosphonates improve BMD by 30% over 2 years
- Reduced fracture incidence with treatment
- Vertebral fractures respond well to conservative management
Outcome Summary
| Condition | Outcome | Key Factor |
|---|---|---|
| Vertebral fractures | Good with bisphosphonates | Pain control, prevent new fractures |
| Long bone fractures | Delayed healing common | Protected weight-bearing |
| EMH cord compression | Good with early treatment | Transfusion + radiation |
| Osteoporosis | Improved with treatment | Multifactorial management |
Controversies & Areas of Uncertainty
Pharmacological choice for osteoporosis - Bisphosphonates (alendronate, zoledronic acid) and denosumab all increase BMD in randomized trials, but no head-to-head fracture-endpoint trial exists in thalassaemia. Concerns over bisphosphonate retention and rebound bone loss after denosumab withdrawal are extrapolated from the general population and remain unresolved in this young, lifelong-treatment cohort.
Duration and safety of long-term therapy - Patients often need decades of anti-resorptive therapy starting in adolescence. The risks of atypical femoral fracture and osteonecrosis of the jaw with prolonged exposure, and the role of drug holidays, are not defined for thalassaemia.
Role of bone-forming agents - Teriparatide and sclerostin antibodies (romosozumab) are theoretically attractive given low bone formation, but evidence in thalassaemia is minimal and they are not yet recommended.
EMH cord compression - first-line treatment - Transfusion, radiotherapy, hydroxyurea and surgical decompression are all used, but the optimal sequence is debated. Many advocate transfusion plus radiotherapy first, reserving surgery for rapidly progressive deficit; high-quality comparative data are lacking because the condition is rare.
Curative therapy and bone outcomes - Allogeneic transplant and gene therapy (e.g. beta-globin lentiviral and gene-editing approaches) can render patients transfusion-independent, but whether established skeletal disease and osteoporosis fully reverse, and long-term bone outcomes, remain uncertain.
Examiner Tip
If asked "which drug?", state that bisphosphonates and denosumab both have RCT-level BMD evidence in thalassaemia, then acknowledge the absence of fracture-endpoint and head-to-head data and the open question of treatment duration in young patients. Demonstrating awareness of the uncertainty scores higher than naming a single agent.
Evidence Base
- Osteoporosis is a major cause of morbidity in adult thalassaemia major
- Pathogenesis is multifactorial: marrow expansion, endocrine dysfunction, iron overload, COLIA1 polymorphism
- RANK/RANKL/OPG pathway is the dominant final mediator of increased osteoclast activity
- Bisphosphonates (potent osteoclast inhibitors) give encouraging results
- Single-centre randomized placebo-controlled trial, 66 thalassaemia patients with osteoporosis
- Zoledronic acid 4 mg IV every 3 months significantly increased lumbar spine BMD at 12 months
- Marked reduction in bone pain and resorption markers (CTX); placebo group worsened
- No BMD gain with the every-6-month schedule
- Randomized, double-blind, placebo-controlled phase 2b trial (n=63; NCT02559648)
- Denosumab 60 mg SC at day 0 and 180 raised lumbar spine BMD 5.92% vs 2.92% placebo (p=0.043)
- Wrist BMD and pain scores improved; sRANKL and resorption markers fell significantly
- No grade 3-4 toxicity
- 2-year randomized placebo-controlled trial in 25 young beta-thalassaemia major patients (mean age 26.6 years)
- Daily oral alendronate significantly increased lumbar and femoral neck BMD vs placebo
- Intramuscular clodronate was ineffective at the dose used
- Lumbar BMD fell significantly in the placebo group
- Cross-sectional study of 111 optimally transfused and chelated adults (mean age 32.6 years)
- Bone demineralisation in 92.7% despite best care; osteopenia at femur, osteoporosis at lumbar spine
- Hypogonadism lowered femoral T-score even when hormonally replaced
- Low BMI, low alkaline phosphatase and hypoparathyroidism predicted worse bone mass
- Taiwanese nationwide cohort: 1369 transfusion-naive thalassaemia subjects vs 5416 matched controls
- 1.35-fold higher overall fracture risk after adjustment for age, sex and comorbidities
- 1.46-fold higher risk of upper-limb fracture; risk most evident in males
- Recurrent spinal epidural extramedullary haematopoiesis causing cord compression in beta-thalassaemia major
- Transfusion reduces the erythropoietic drive sustaining EMH
- Combination of surgery and radiotherapy gave complete resolution at 2-year follow-up
- Pre-transfusion haemoglobin target 9-10.5 g/dL to suppress ineffective erythropoiesis and marrow expansion
- Annual DEXA from adolescence; correct vitamin D, calcium and hypogonadism
- Bisphosphonates are the recommended pharmacotherapy for established osteoporosis
Viva Scenarios
Use these scenarios to practise clinical reasoning and management decisions
Vertebral Fracture in Young Thalassemia Patient
"18-year-old male with beta thalassemia major on regular transfusions presents with 3 weeks of progressively worsening mid-back pain. X-ray shows T12 compression fracture with 40% height loss. How do you assess and manage this patient?"
This is a **pathological vertebral compression fracture** secondary to **thalassemia-related osteoporosis**. My assessment and management would be:
Assessment:
- Full history: Trauma history (likely minimal), duration and character of pain, neurological symptoms
- Examination: Neurological examination of lower limbs, kyphotic deformity
- DEXA scan to quantify bone density
- MRI spine if neurological symptoms or to assess fracture acuity
- Laboratory: Vitamin D, calcium, endocrine panel (hypogonadism screen)
Management:
- Analgesia and activity modification
- Thoracolumbosacral orthosis (TLSO) for comfort and support
- Start bisphosphonate therapy (e.g., zoledronic acid 4mg IV)
- Optimize vitamin D (target greater than 75 nmol/L) and calcium
- Hormone replacement if hypogonadal
- Consider vertebroplasty if pain refractory to conservative measures
- Liaise with hematology regarding transfusion and chelation optimization
Paraparesis in Thalassemia - Spinal Cord Compression
"32-year-old female with beta thalassemia intermedia presents with progressive bilateral lower limb weakness and urinary retention over 2 weeks. She is not on regular transfusions. MRI shows a paraspinal mass at T6-T8 causing cord compression. What is your diagnosis and management?"
This is **spinal cord compression from extramedullary hematopoiesis (EMH)**. This is a known complication of thalassemia, particularly intermedia where transfusions are insufficient to suppress marrow expansion.
Immediate Management:
- Urgent hematology consultation
- Blood transfusion to suppress erythropoietic drive - this often leads to rapid improvement
- High-dose corticosteroids (dexamethasone 10mg stat then 4mg QID)
- Monitor neurological status closely
Definitive Treatment Options:
- Radiation therapy: EMH is very radiosensitive (low doses effective)
- Hydroxyurea: Reduces EMH by increasing HbF
- Surgical decompression: If rapidly progressive or failure of conservative measures
- Initiate regular transfusion program to prevent recurrence
Surgical Considerations:
- EMH tissue is highly vascular - risk of significant bleeding
- Coordinate with hematology for preoperative transfusion
- Complete resection not always necessary - decompression is goal
- Radiation often used as adjunct
Hair-on-end Skull in Child
"You are shown a skull X-ray of a 7-year-old with a classic 'hair-on-end' appearance. The child is from Southeast Asia and has pallor and splenomegaly. What is your differential diagnosis and approach?"
The **hair-on-end appearance** on skull X-ray is classic for **chronic hemolytic anemia with marrow expansion**. In this clinical context, **thalassemia major** is the most likely diagnosis.
Differential Diagnosis:
- Thalassemia major (most likely given geography)
- Sickle cell disease (less common in Southeast Asia)
- Hereditary spherocytosis
- Pyruvate kinase deficiency
- Severe iron deficiency anemia (rare cause)
Approach:
- CBC: Expect severe microcytic anemia
- Blood smear: Target cells, nucleated RBCs, basophilic stippling
- Hemoglobin electrophoresis: Increased HbF, absent HbA in beta major
- Family history and genetic testing if needed
Management Implications:
- If thalassemia major confirmed: Needs regular transfusion program
- Early transfusions prevent skeletal deformities
- Chelation therapy to prevent iron overload
- Bone marrow transplant consideration if suitable donor
MCQ Practice Points
Classic Radiographic Finding
Q: What is the pathognomonic skull radiograph finding in thalassemia major, and why is the occipital region spared?
A: Hair-on-end (crew-cut) appearance from diploic expansion due to marrow hyperplasia. The occipital region is spared because it contains minimal marrow (predominantly diploe only). This finding occurs due to chronic erythroid hyperplasia compensating for hemolytic anemia.
Osteoporosis Mechanisms
Q: What are the four main mechanisms of osteoporosis in thalassemia patients?
A: Marrow expansion (cortical thinning from erythroid hyperplasia), iron toxicity (direct osteoblast inhibition from transfusion overload), deferoxamine toxicity (chelation therapy inhibits osteoblast function), and hypogonadism (iron deposition in pituitary causes hormonal deficiency). This is why 40-80% of adults have osteoporosis.
Extramedullary Hematopoiesis
Q: What is the orthopaedic emergency associated with extramedullary hematopoiesis in thalassemia?
A: Spinal cord compression. Paraspinal extramedullary hematopoietic tissue can expand and compress the cord, typically in the thoracic region. Treatment includes urgent hypertransfusion (suppresses marrow), radiation therapy, and surgical decompression if severe. MRI shows characteristic paraspinal masses with T1/T2 intermediate signal.
Inheritance Pattern
Q: A 3-year-old from the Mediterranean region presents with severe anemia, hepatosplenomegaly, and frontal bossing. Parents are asymptomatic. What is the inheritance pattern?
A: Autosomal recessive. Beta thalassemia major requires inheritance of two defective beta-globin alleles. Parents are carriers (thalassemia minor) and typically asymptomatic with mild microcytic anemia. Mediterranean, Middle Eastern, and Southeast Asian populations have high carrier frequencies due to malaria protection.
Guidelines, Registries & Global Practice
Global Epidemiology
Burden and Distribution:
- Thalassaemia is the most common monogenic disorder worldwide; roughly 5-7% of the global population carry a haemoglobinopathy gene
- Approximately 60,000-70,000 children are born with a severe thalassaemia each year, concentrated in the Mediterranean, Middle East, South Asia and Southeast Asia ("thalassaemia belt")
- Carrier frequency reaches 10-30% in highly endemic regions
- High carrier rates reflect the heterozygote survival advantage against falciparum malaria
- Migration has made thalassaemia a clinically relevant diagnosis in Northern Europe, North America and Australasia
Service Model (resource-rich settings):
- Comprehensive thalassaemia centres delivering multidisciplinary care (haematology, endocrinology, cardiology, orthopaedics)
- National blood services for safe, leucodepleted transfusion supply
- Genetic counselling and antenatal/carrier screening programmes
THALASSEMIA
Clinical summary
PATHOLOGY
- •Globin chain synthesis defect (quantity)
- •Beta major: Transfusion-dependent from infancy
- •Autosomal recessive inheritance
- •Mediterranean, Middle East, SE Asia
SKELETAL CHANGES
- •Hair-on-end skull (occipital spared)
- •Chipmunk facies (maxillary expansion)
- •Widened medullary cavities
- •Cortical thinning
- •Osteoporosis (40-80%)
OSTEOPOROSIS CAUSES - MIDHE
- •Marrow expansion
- •Iron overload (toxic to osteoblasts)
- •Deferoxamine (chelation effect)
- •Hypogonadism
- •Endocrine (vitamin D, thyroid)
MANAGEMENT
- •Transfusions + chelation (hematology)
- •DEXA screening for osteoporosis
- •Bisphosphonates (zoledronic acid)
- •Vitamin D and calcium
- •Vertebroplasty for refractory pain
EMH CORD COMPRESSION
- •Transfusion first (reduces EMH)
- •Radiation (EMH is radiosensitive)
- •Surgery if rapidly progressive
- •Multidisciplinary management