Melanoma of Soft Parts | EWSR1-ATF1 Fusion | Young Adults | Distal Extremities
Defining Features
Critical Must-Knows
- Clear cell sarcoma is the melanoma of soft parts - it shows melanocytic differentiation but is NOT cutaneous melanoma
- EWSR1-ATF1 fusion from t(12;22) is the molecular hallmark and is ABSENT in true melanoma - this is the key discriminator
- Most common around the foot and ankle, attached to tendons and aponeuroses, in young adults
- High rate of lymph node spread (around 15-20%) - unusual for a soft tissue sarcoma
- Chemotherapy and radiotherapy are largely ineffective - complete wide surgical excision is the mainstay
Clinical Pearls
- "A melanocytic-looking deep tumour near a tendon with NO skin lesion is clear cell sarcoma until molecular testing proves otherwise
- "EWSR1 break-apart FISH or RT-PCR separates clear cell sarcoma (positive) from melanoma (negative)
- "Late metastases beyond 10 years occur, so lifelong surveillance is needed
- "Even small tumours can metastasise, so size never makes the lesion safe
Clinical Imaging
Imaging Gallery
Critical Clear Cell Sarcoma Exam Points
It Is NOT Melanoma
Clear cell sarcoma looks like melanoma but is a distinct sarcoma. It shares melanocytic markers (S100, HMB-45, Melan-A, SOX10) but carries the EWSR1-ATF1 fusion that true melanoma lacks. Always confirm with molecular testing and exclude a primary skin melanoma.
Surgery Is the Treatment
Wide complete excision is the only reliably effective treatment. Chemotherapy and radiotherapy have limited activity. An incomplete (R+) resection markedly worsens survival, so plan the definitive operation carefully.
Lymph Node Spread
Nodal metastasis is relatively common (around 15-20%) - unusual for a soft tissue sarcoma. Examine regional nodes and consider sentinel lymph node biopsy as part of staging.
Small Does Not Mean Safe
Even small tumours metastasise. Slow growth and small size are reassuring in many lesions but not here. Late relapse beyond 10 years means surveillance must be long term.
MELANOMAClear Cell Sarcoma Key Features
| M | Melanocytic markers S100, HMB-45, Melan-A, SOX10 positive |
| E | EWSR1-ATF1 fusion t(12;22) defining hallmark, absent in melanoma |
| L | Lymph node spread Around 15-20%, unusual for soft tissue sarcoma |
| A | Aponeuroses and tendons Arises from deep tendinous tissue |
| N | Not cutaneous melanoma Distinct sarcoma, no overlying skin lesion |
| O | Of soft parts Historic name melanoma of soft parts |
| M | Mainstay is surgery Wide excision, chemo and radiation poorly effective |
| A | Ankle and foot Most common location, young adults |
| M | Melanocytic markers S100, HMB-45, Melan-A, SOX10 positive | A | Aponeuroses and tendons Arises from deep tendinous tissue | M | Mainstay is surgery Wide excision, chemo and radiation poorly effective |
| E | EWSR1-ATF1 fusion t(12;22) defining hallmark, absent in melanoma | N | Not cutaneous melanoma Distinct sarcoma, no overlying skin lesion | A | Ankle and foot Most common location, young adults |
| L | Lymph node spread Around 15-20%, unusual for soft tissue sarcoma | O | Of soft parts Historic name melanoma of soft parts |
Hook:MELANOMA spells out the look-alike that clear cell sarcoma is NOT - the fusion gene is the giveaway!
FUSEDistinguishing Clear Cell Sarcoma from Melanoma
| F | Fusion gene present EWSR1-ATF1 in clear cell sarcoma, absent in melanoma |
| U | Underlying skin normal No primary cutaneous or mucosal melanoma lesion |
| S | Site is deep soft tissue Tendons and aponeuroses, not epidermis |
| E | Extremity of young adult Foot, ankle and knee in patients 20-40 years |
| F | Fusion gene present EWSR1-ATF1 in clear cell sarcoma, absent in melanoma | S | Site is deep soft tissue Tendons and aponeuroses, not epidermis |
| U | Underlying skin normal No primary cutaneous or mucosal melanoma lesion | E | Extremity of young adult Foot, ankle and knee in patients 20-40 years |
Hook:The fusion gene FUSEs the diagnosis together - melanoma cannot FUSE!
MILDPoor Prognostic Factors in Clear Cell Sarcoma
| M | Metastases at diagnosis Distant disease confers dismal survival |
| I | Incomplete resection (R+) Positive margins greatly reduce survival |
| L | Large tumour size Larger tumours carry worse outcomes |
| D | Deep and necrotic Deep location and necrosis indicate aggression |
| M | Metastases at diagnosis Distant disease confers dismal survival | L | Large tumour size Larger tumours carry worse outcomes |
| I | Incomplete resection (R+) Positive margins greatly reduce survival | D | Deep and necrotic Deep location and necrosis indicate aggression |
Hook:MILD outcomes are anything but mild when these factors are present!
Overview and Epidemiology
Clear cell sarcoma (CCS) is a rare, highly malignant soft tissue sarcoma that arises from tendons and aponeuroses, classically in the distal extremities of young adults. It was historically called malignant melanoma of soft parts because its cells share melanocytic differentiation with cutaneous melanoma. However, modern molecular pathology shows it is a distinct entity defined by the EWSR1-ATF1 gene fusion from the t(12;22)(q13;q12) translocation, which is absent in true melanoma.
A Misleading Name
Clear cell sarcoma is the great melanoma mimic. The cells express S100, HMB-45, Melan-A and SOX10, can contain melanin, and can spread to lymph nodes like melanoma. The decisive difference is the EWSR1-ATF1 fusion gene, which is present in clear cell sarcoma and absent in melanoma. In the exam, if you are shown a deep soft tissue tumour with melanocytic markers but no skin lesion, think clear cell sarcoma and ask for molecular testing.
Demographics
- Frequency: About 1% of all soft tissue sarcomas (very rare)
- Age: Peak in young adults, roughly 20 to 40 years
- Sex: Roughly equal, slight female predominance in some series
- Paediatric: Occurs in children but is uncommon
Location Distribution
- Foot and ankle: Most common site, attached to tendons
- Knee and lower leg: Frequent
- Hand, wrist and forearm: Less common but reported
- Trunk and head/neck: Rare
Behaviour and Spread
| Feature | Clear Cell Sarcoma | Exam Relevance |
|---|---|---|
| Growth | Slow-growing deep mass, often present for months to years | Long history can falsely reassure - delayed diagnosis is common |
| Lymph node spread | Relatively common (around 15-20%) | Examine nodes and consider sentinel node biopsy |
| Distant metastases | Lung most common, then bone | CT chest for staging, late relapse beyond 10 years |
| Chemosensitivity | Low - conventional chemotherapy largely ineffective | Do not rely on systemic therapy for cure |
Pathophysiology and Molecular Biology
Cellular Origin and Genetics
Clear cell sarcoma is thought to arise from neural crest-derived cells with the capacity for melanocytic differentiation. This explains the melanocytic markers and occasional melanin production. The oncogenic driver is a fusion gene that acts as an aberrant transcription factor and switches on the melanocyte master regulator MITF.
EWSR1-ATF1 Fusion
The defining molecular hallmark, present in the large majority of cases:
- Translocation t(12;22)(q13;q12)
- Fuses EWSR1 on chromosome 22 with ATF1 on chromosome 12
- Creates an aberrant transcription factor that activates the MITF melanocytic program
- Detected by EWSR1 break-apart FISH or RT-PCR
Variant Fusion and Relatives
A minority carry EWSR1-CREB1:
- EWSR1-CREB1 is a less common alternative fusion
- The gastrointestinal counterpart is the malignant gastrointestinal neuroectodermal tumour (GNET), which shares EWSR1 rearrangement but lacks melanocytic marker expression
- PRAME immunostaining can support the diagnosis but does not separate CCS from melanoma alone
Molecular Testing Resolves the Diagnosis
EWSR1 rearrangement testing is the key to diagnosis. Because clear cell sarcoma and melanoma share melanocytic immunohistochemistry, a melanocytic-appearing deep soft tissue tumour must be tested for the EWSR1-ATF1 (or EWSR1-CREB1) fusion. A positive fusion confirms clear cell sarcoma; melanoma is fusion-negative. Always exclude a cutaneous or mucosal primary melanoma on clinical grounds as well.
Classification and Histology
Histological and Immunohistochemical Features
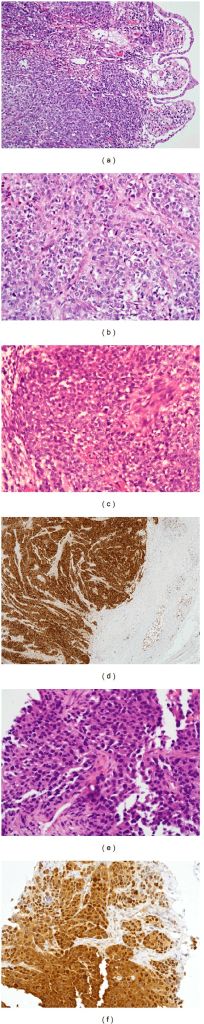
Morphology
Clear cell sarcoma has a distinctive architecture that, combined with markers and molecular testing, secures the diagnosis.
Architecture
- Nests and fascicles of cells separated by fibrous septa
- Cells are epithelioid to spindled
- Pale or clear cytoplasm (glycogen-rich)
- Prominent nucleoli, scattered wreath-like multinucleated giant cells
Other Features
- Melanin pigment present in a minority of cases
- Low to moderate mitotic activity
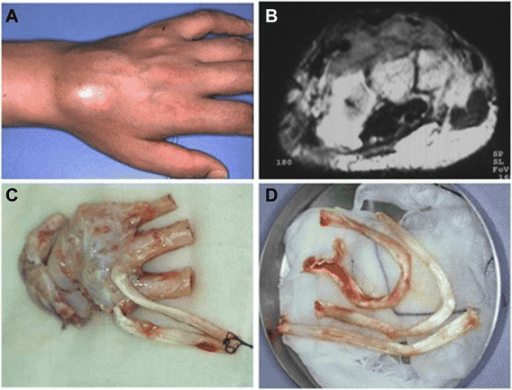
- Attachment to tendon or aponeurosis on resection
- Necrosis indicates more aggressive disease
Clinical Presentation
History and Examination
Presenting Symptoms
- Slow-growing deep mass, often around the foot, ankle or knee
- Frequently painless initially, pain with growth or nerve involvement
- Long duration (months to years) is common
- Attachment to tendon may cause mechanical symptoms
Examination
- Firm deep mass, often fixed to deep structures
- Regional lymph nodes must be examined (nodal spread relatively common)
- No overlying skin lesion (helps exclude cutaneous melanoma)
- Document neurovascular status distal to the mass
Diagnostic Delay Is Common
The slow growth of clear cell sarcoma frequently leads to delay or to it being mistaken for a benign lesion. Many tumours are removed by an unplanned excisional biopsy because a benign cause is assumed. Any persistent deep soft tissue mass in a young adult, especially near a tendon, should be imaged and biopsied at a sarcoma centre before excision.
Investigations and Imaging
Imaging Protocol
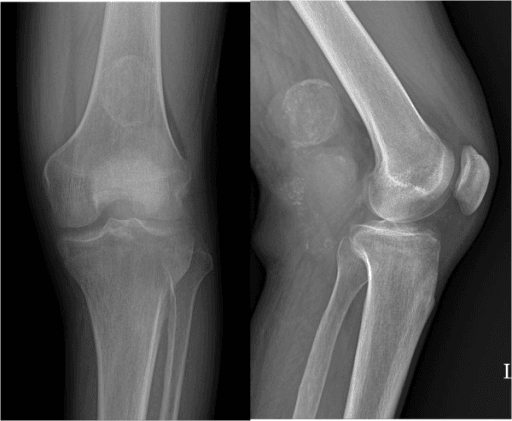
MRI (Local Staging)
MRI of the whole compartment is the key local staging investigation.
- T1: Clear cell sarcoma is often slightly high signal on T1 due to melanin (a useful but inconstant clue)
- T2: Variable, usually intermediate to high signal
- Post-contrast: Enhancement, often heterogeneous
- Define relationship to tendons, neurovascular bundle and joint
T1 Hyperintensity Clue
Melanin within clear cell sarcoma can produce high T1 signal, similar to melanoma. This is a helpful pointer in a deep tendinous mass but is not always present and never replaces biopsy and molecular testing.
Biopsy
Biopsy Principles
Plan the biopsy with the definitive surgeon at a sarcoma centre.
- Core needle biopsy is preferred, with the tract placed so it can be excised at surgery
- Avoid unplanned excisional biopsy - it is common in clear cell sarcoma because a benign lesion is assumed, and it compromises margins
- Request EWSR1 fusion testing (break-apart FISH or RT-PCR) on the specimen
- Send adequate tissue for melanocytic immunohistochemistry and molecular studies
Staging System
AJCC 8th Edition Soft Tissue Sarcoma (Extremity/Trunk) - principles
| Stage | Grade | Size/Node/Metastasis | Implication |
|---|---|---|---|
| IA / IB | Low | Small or larger but low grade, N0 M0 | Better outlook, surgery centred |
| II / IIIA | High | Up to 10cm, N0 M0 | High-grade local disease, plan wide excision |
| IIIB | High | Greater than 10cm, N0 M0 | Larger high-grade tumour, worse prognosis |
| IV | Any | Node positive (N1) or distant metastasis (M1) | Nodal disease upstages to IV in this system |
Nodal Disease Matters
In the AJCC 8th edition soft tissue sarcoma system, regional node-positive disease is classified as stage IV. This is especially relevant in clear cell sarcoma because of its relatively high rate of lymph node spread.
Management
Core Principles
Surgery Is the Mainstay
Complete wide surgical excision with negative margins is the only reliably effective treatment for clear cell sarcoma. Chemotherapy is largely ineffective and radiotherapy has only a limited adjuvant role. The single most important predictor of survival in localised disease is achieving an R0 (microscopically clear) resection.
Treatment fundamentals:
- Surgery: Wide excision with a cuff of normal tissue; amputation only if limb salvage cannot achieve clear margins
- Lymph nodes: Assess regional nodes; sentinel node biopsy often considered, therapeutic lymphadenectomy for proven nodal disease
- Radiotherapy: Considered for close or positive margins or large high-grade tumours
- Chemotherapy: Limited efficacy; reserved for selected advanced cases within trials
Prognosis and Outcomes
Prognostic Factors
| Factor | Favourable | Unfavourable |
|---|---|---|
| Tumour size | Small | Large |
| Margins | R0 (clear) | R+ (positive) |
| Metastases | None at diagnosis | Nodal or distant at diagnosis |
| Necrosis | Absent | Present |
| Depth | More superficial | Deep |
Survival Overview
Approximate Survival
| Category | 5-Year Survival | Comment |
|---|---|---|
| Localised, completely resected | Around 50-65% | Best outcome group |
| Overall (all comers) | Around 40-60% | Falls with longer follow-up |
| Positive margins (localised) | Markedly reduced | R+ strongly worsens survival |
| Metastatic at diagnosis | Very poor (around 1-2 years median) | Largely incurable |
Late Relapse
Clear cell sarcoma can recur or metastasise many years after treatment, sometimes beyond 10 years. Long-term surveillance with clinical examination, chest imaging and regional node assessment is essential, and apparent early cure does not guarantee long-term cure.
Clinical Relevance and Surveillance
Why This Matters in the Exam and Clinic
Avoid the Unplanned Excision
Because the tumour is slow-growing and often assumed benign, it is frequently removed by an unplanned excision that leaves positive margins. Recognising the entity and referring before biopsy preserves the chance of an R0 resection, which is the main driver of survival.
Surveillance Strategy
- Clinical examination including regional nodes
- Chest imaging for lung metastases
- Local MRI if recurrence suspected
- Continue surveillance long term given late relapse
Safe-Practice Points
Applicable in any health system:
- Refer any persistent deep soft tissue mass to a sarcoma centre before biopsy
- Confirm molecular testing for the EWSR1 fusion was requested
- Assess and document regional lymph nodes
- Counsel on the need for complete surgery, the limited role of chemo and radiotherapy, and the risk of late relapse
- Provide a written long-term surveillance plan
Evidence Base and Key Studies
Largest Modern Cohort: Prognostic Factors in Clear Cell Sarcoma
- Single-centre retrospective series of 43 patients with clear cell sarcoma and the GINET variant, one of the largest clinical cohorts reported
- EWSR1::ATF1 translocation positive in 24 of 26 tissues tested (94%); median tumour size only 3.6 cm
- Complete R0 resection correlated significantly with survival in localised disease (5-year overall survival 64% with R0 versus 0% with R+, p=0.017)
- Lymphatic spread in 18.6% and distant metastases in 20.9%; initial metastatic disease gave a dismal median survival of 1.4 years versus 7.1 years for M0 (p less than 0.001)
Aggressive Behaviour and Chemoresistance in Paediatric Clear Cell Sarcoma
- Cooperative-group series of 28 paediatric patients treated 1980-2000 by the Italian and German soft tissue sarcoma groups
- 5-year overall and event-free survival of 66.4% and 63.3% after a median follow-up of 102 months
- Only 1 partial and 1 minor response among 7 chemotherapy-evaluable patients, with 5 showing no response
- Outcome differed significantly by IRS group, tumour size and site; complete resection was the mainstay and the only treatment needed for small tumours
EWSR1 Break-Apart FISH Distinguishes Clear Cell Sarcoma from Melanoma
- Eighteen non-cutaneous melanocytic tumours examined with EWSR1 (22q12) break-apart FISH and RT-PCR
- Two cases were reclassified as clear cell sarcoma based on EWSR1 rearrangement (mean 67.5% positive cells)
- Type 1 EWSR1 exon 8-ATF1 exon 4 fusion transcripts confirmed in the FISH-positive cases by RT-PCR
- The remaining 16 melanomas lacked EWSR1 break-apart signals
EWSR1-ATF1 Fusion and the Role of PRAME (Narrative Review)
- Synthesises that clear cell sarcoma arises from connective tissue and is defined by the EWSR1-ATF1 fusion from t(12;22), absent in melanoma
- Highlights PRAME as an immunohistochemical marker that supports the diagnosis but does not by itself separate clear cell sarcoma from melanoma
- Wide surgical excision is the cornerstone for localised disease, with sentinel lymph node biopsy aiding staging and adjuvant radiotherapy in selected cases
- Chemotherapy has limited efficacy; targeted therapy against EWSR1-ATF1-driven pathways and immune checkpoint inhibitors are emerging strategies
Lymph Node Metastasis Across Soft Tissue Sarcomas (SEER)
- SEER analysis of 15,525 adults with soft tissue sarcoma diagnosed 2004-2013
- Overall lymph node metastasis at diagnosis was 5.3%, but clear cell sarcoma was among the subtypes with the highest nodal rates (alongside rhabdomyosarcoma, epithelioid and myxoid/round cell liposarcoma)
- Lymph node metastasis independently predicted worse overall survival (HR 1.34, p less than 0.001)
- Among node-positive patients, lymphadenectomy combined with limb salvage was associated with the highest 5-year survival (HR 0.46)
Sunitinib Activity in Clear Cell Sarcoma
- Report of objective tumour response to the multitargeted tyrosine kinase inhibitor sunitinib in clear cell sarcoma
- Provides clinical proof of principle that targeting kinase pathways (including MET signalling downstream of the fusion-driven MITF program) can produce responses
- Relevant to a tumour type that is largely resistant to conventional cytotoxic chemotherapy
- Supports investigating molecularly targeted agents in advanced clear cell sarcoma
Exam Viva Scenarios
Use these scenarios to practise clinical reasoning and management decisions
Scenario 1: Young Adult with a Foot Mass - Diagnosis
"A 27-year-old man presents with a 3cm firm, slow-growing mass on the dorsum of the foot, present for about 2 years and attached to the deep tissues near the extensor tendons. There is no overlying skin lesion. How would you investigate, and what diagnosis must you not miss?"
Scenario 2: Distinguishing Clear Cell Sarcoma from Melanoma
"A pathologist reports a deep soft tissue tumour that is S100, HMB-45 and Melan-A positive and asks whether this is metastatic melanoma or clear cell sarcoma. There is no known skin primary. How do you resolve this?"
Scenario 3: The Already-Excised Lesion
"A 30-year-old woman had a 2.5cm mass marginally excised from the ankle as a presumed ganglion. Histology returns as clear cell sarcoma with positive margins. CT chest is clear. How do you manage her now?"
MCQ Practice Points
Molecular Biology Question
Q: What gene fusion defines clear cell sarcoma and distinguishes it from melanoma? A: The EWSR1-ATF1 fusion from t(12;22)(q13;q12) (with EWSR1-CREB1 as a less common variant). It is present in clear cell sarcoma and absent in melanoma, making EWSR1 break-apart FISH or RT-PCR the key discriminator.
Diagnosis Question
Q: Why can immunohistochemistry not separate clear cell sarcoma from melanoma? A: Both express the same melanocytic markers (S100, HMB-45, Melan-A, SOX10). Only molecular testing for the EWSR1 fusion reliably distinguishes them.
Location Question
Q: Where does clear cell sarcoma most commonly arise? A: In the deep soft tissue of the distal extremities, especially the foot and ankle, attached to tendons and aponeuroses, in young adults aged roughly 20 to 40 years.
Treatment Question
Q: What is the mainstay of treatment for localised clear cell sarcoma? A: Complete wide surgical excision with negative (R0) margins. Chemotherapy is largely ineffective and radiotherapy has a limited adjuvant role. R0 resection is the strongest predictor of survival.
Spread Question
Q: What pattern of spread is unusually common in clear cell sarcoma for a soft tissue sarcoma? A: Lymph node metastasis (around 15-20%), in addition to lung and bone, which is why regional nodes should be assessed and sentinel node biopsy considered.
Guidelines, Registries & Global Practice
Global Epidemiology
Incidence and Burden
- Rarity: About 1% of all soft tissue sarcomas worldwide
- Age: Predominantly young adults (peak roughly 20 to 40 years)
- Site: Strong predilection for the distal lower limb, especially foot and ankle
- Variant: The gastrointestinal counterpart (GINET) shares the EWSR1 rearrangement
Outcome Data
- Nodal spread: Among the highest of soft tissue sarcomas (SEER data)
- Metastatic pattern: Lung dominant, then bone and lymph nodes
- Survival: Roughly 40 to 65% at 5 years, driven heavily by completeness of resection
- Late relapse: Recurrence beyond 10 years is well recognised
Side-by-Side Guidance from Major Bodies
| Body | Diagnosis | Local Treatment | Systemic Therapy |
|---|---|---|---|
| ESMO/EURACAN (Europe) | Refer to sarcoma centre before biopsy; molecular confirmation of EWSR1 fusion | Wide excision with radiotherapy for high-risk tumours | Conventional chemotherapy of limited value; targeted/trial options for advanced disease |
| NCCN (US) | Core biopsy at a sarcoma centre; multidisciplinary review and node assessment | Limb-sparing wide excision; re-excision for positive margins | Individualised systemic therapy; tyrosine kinase inhibitors considered in relapse |
| NICE / BSG sarcoma (UK) | Mandatory specialist sarcoma MDT before definitive treatment | Centralised surgery and radiotherapy at designated centres | Chemotherapy reserved for selected advanced cases |
Universal Principle Across Guidelines
Across regions the agreed principles are the same: refer any suspicious deep or enlarging soft tissue mass to a specialist sarcoma centre before biopsy, confirm the diagnosis molecularly (EWSR1 fusion), and plan definitive treatment through a multidisciplinary sarcoma team centred on complete surgical excision.
High- vs Limited-Resource Practice Variation
High-Resource Settings
- Routine EWSR1 break-apart FISH, RT-PCR or NGS for fusion confirmation
- Centralised sarcoma MDTs with limb-salvage surgery and reconstruction
- Sentinel lymph node biopsy and PET-CT staging
- Access to targeted agents and clinical trials for advanced disease
Limited-Resource Settings
- Diagnosis may rely on morphology plus melanocytic immunohistochemistry where molecular testing is unavailable
- Later presentation with larger tumours and higher amputation rates
- Restricted radiotherapy and targeted-therapy access
- Telepathology and regional referral networks help bridge gaps
CLEAR CELL SARCOMA
Clinical summary
Key Identity
- •Rare soft tissue sarcoma (about 1% of all soft tissue sarcomas)
- •Historic name: malignant melanoma of soft parts
- •Young adults, deep distal extremities, especially foot and ankle, on tendons and aponeuroses
- •Shows melanocytic differentiation but is NOT cutaneous melanoma
Molecular Biology (Defining)
- •EWSR1-ATF1 fusion from t(12;22)(q13;q12) is the hallmark
- •EWSR1-CREB1 is a less common variant fusion
- •Fusion is ABSENT in melanoma - the key discriminator
- •Detect with EWSR1 break-apart FISH or RT-PCR; fusion drives MITF and MET
Pathology and Markers
- •Nests of epithelioid to spindled cells with clear cytoplasm and wreath-like giant cells
- •Positive for S100, HMB-45, Melan-A, SOX10 (shared with melanoma)
- •PRAME often positive but does not separate CCS from melanoma
- •Differentials: melanoma, MPNST, synovial sarcoma, PEComa, GNET
Clinical and Imaging
- •Slow-growing deep mass, often painless, long history, diagnostic delay common
- •MRI may show high T1 signal from melanin; define compartment and tendons
- •CT chest for lung metastases; assess regional nodes, consider sentinel node biopsy
- •Node-positive disease is stage IV in the AJCC system
Management
- •Complete wide surgical excision with R0 margins is the mainstay
- •Refer before biopsy; avoid unplanned excisional biopsy
- •Radiotherapy for close/positive margins or large high-grade tumours
- •Conventional chemotherapy largely ineffective; tyrosine kinase inhibitors (e.g. sunitinib) in advanced disease/trials
Prognosis and Surveillance
- •R0 resection is the strongest survival predictor (R+ markedly worse)
- •Even small tumours metastasise; lung then bone and nodes
- •5-year survival roughly 40-65%, falling with longer follow-up
- •Late relapse beyond 10 years - long-term surveillance is essential