SLC26A2 Sulfate Transporter Defect | Short-Limbed Dwarfism | Hitchhiker Thumb + Cauliflower Ear
- Autosomal recessive - both parents carriers, recurrence risk 1 in 4
- SLC26A2 (formerly DTDST) - sulfate-chloride antiporter; cartilage proteoglycan undersulfation
- Hitchhiker thumb = abducted, hypermobile, proximally inserted thumb - pathognomonic
- Cauliflower ear (cystic pinnae) appears in first weeks of life - diagnostic
- Rigid clubfoot resists Ponseti - often needs posteromedial release
- Cervical kyphosis at C3-C5 - usually self-corrects; brace if progressive
- Progressive scoliosis begins in early childhood - long fusion often required
- Intelligence and life expectancy are NORMAL - rehab focus is mobility
- “DTDST = Diastrophic Dysplasia Sulfate Transporter = same gene as SLC26A2
- “Cauliflower ear is present at birth or within weeks - pathognomonic
- “Hitchhiker thumb = 'hitching a ride' - abduction deformity, NOT absent thumb
- “Clubfoot is rigid and resists standard Ponseti casting
- “Spine and hip surgery carry high complication rates (small bones, soft tissue)
- “Final adult height often 100-140 cm (short-limbed dwarfism)
SLC26A2 (Solute Carrier 26, family A, member 2) - originally called DTDST. Autosomal recessive sulfate-chloride antiporter on chromosome 5q32-q33.1. Loss of function leads to undersulfation of cartilage proteoglycans - chondrocytes cannot incorporate sulfate, leading to abnormal collagen matrix and disorganised growth plate.
Hitchhiker thumb + cauliflower ear + micromelia + rigid clubfoot = classic tetrad. The cystic swelling of the pinnae (cauliflower ear) appears within the first weeks of life and is essentially diagnostic in a short-limbed newborn.
Cervicothoracic kyphosis at C3-C5 is present at birth in 30-50 percent. Most resolve spontaneously with neck extension and growth. Persistent or progressive kyphosis (greater than 60 degrees) needs MRI + possible posterior fusion.
Cervical spine instability + small mouth + short neck = difficult intubation. ALWAYS obtain lateral cervical spine X-ray + flexion-extension views before intubation. Micrognathia and cleft palate also common (30 percent).
- Diagnosis
- Clinical + SLC26A2 sequencing, X-ray survey
- Treatment
- Counselling, multi-disciplinary surveillance
- Key Pearl
- Cauliflower ear appears in first weeks - diagnostic
- Diagnosis
- Pirani score + clinical exam + X-ray
- Treatment
- Often needs posteromedial soft-tissue release
- Key Pearl
- Recurrence is the norm - serial surgery common
- Diagnosis
- Whole-spine EOS radiograph, MRI of cord
- Treatment
- Brace if 20-40 degrees, fusion if 50+ degrees
- Key Pearl
- Long-segment fusion often needed - high complication rate
- Diagnosis
- Lateral cervical X-ray + flexion-extension MRI
- Treatment
- Posterior cervical fusion ± decompression
- Key Pearl
- Most resolve spontaneously - operate only if progressive
DAASLC26A2 Spectrum - Phenotypic Series
Hook:Same SLC26A2 gene, three phenotypes: DAA = Diastrophic, Atelosteogenesis II, Achondrogenesis 1B
Spine-Hip-Knee-FootSurgical Priorities by Age
Hook:Address the spine, pinnae, hip, knee, and foot in that order to maximise function!
Overview and Epidemiology
Diastrophic dysplasia is the prototype of the SLC26A2 sulfate transporter spectrum - a group of three allelic disorders ranging from perinatally lethal (achondrogenesis 1B) to ambulant short-limbed dwarfism. Diagnosis at birth allows genetic counselling, anticipation of airway and spinal complications, and a multi-stage orthopaedic plan that addresses the cervical spine, scoliosis, hip, knee, clubfoot, and thumb in sequence.
- Incidence: Approximately 1 in 100,000 live births worldwide
- Finnish enrichment: 1 in 33,000 - founder mutation p.Arg279Trp
- No sex predilection - autosomal recessive
- Higher carrier frequency in Finns, French-Canadians, African Americans
- Normal life expectancy with managed cervical and respiratory issues
- Disability: Short stature, restricted joint mobility, pain
- Airway: Cervical instability, micrognathia, cleft palate (30 percent), OSA
- Spine: Cervical kyphosis, progressive scoliosis, cord compression
- Limbs: Hip dysplasia, knee flexion contractures, rigid clubfeet
- Hearing: Recurrent otitis media, mixed hearing loss
Founder effect: The Finnish founder mutation c.862C greater than T (p.Arg279Trp) accounts for 90 percent of Finnish diastrophic dysplasia chromosomes. Other populations have private mutations. Genetic counselling must be informed by population of origin and carrier screening partners from high-prevalence groups.
The topic mentions "prenatal testing options" only in passing, but antenatal diagnosis is genuinely examinable and important for counselling:
- Second-trimester ultrasound can suggest the diagnosis: micromelia (short, bowed long bones) below the expected centile, and - characteristically - the abducted "hitchhiker" thumb and great toe can sometimes be seen held away from the hand/foot. Polyhydramnios (impaired fetal swallowing from micrognathia) is a soft marker, and in the severe allelic forms a narrow thorax signals possible lethality. Diastrophic dysplasia is often first suspected at the routine anomaly scan in an at-risk or even a previously unsuspected pregnancy.
- Molecular prenatal testing is definitive when the family's two SLC26A2 mutations are already known: chorionic villus sampling (around eleven to thirteen weeks) or amniocentesis (from about fifteen weeks) for targeted mutation analysis. For known-carrier couples wishing to avoid an affected pregnancy, preimplantation genetic testing (PGT-M) is an option.
- Counselling: a couple with one affected child are obligate carriers with a one-in-four recurrence risk each pregnancy; the prenatal pathway (and the option of declining testing) should be discussed pre-conceptually.
Exam point: in an at-risk pregnancy, the anomaly-scan triad of micromelia + abducted hitchhiker digits + polyhydramnios should prompt diastrophic dysplasia in the differential, with targeted SLC26A2 CVS/amniocentesis (or PGT-M) giving a definitive answer when the parental mutations are known.
Pathophysiology
The SLC26A2 gene on chromosome 5q32-q33.1 encodes a transmembrane sulfate-chloride (SO4(2-)/Cl-) antiporter that mediates sulfate uptake by chondrocytes in the growth plate. Loss of function reduces intracellular sulfate, leading to undersulfation of cartilage proteoglycans (especially aggrecan). The matrix is mechanically weak, the chondrocytes are disorganised, and the growth plate is structurally abnormal - producing the disproportionate short-limbed dwarfism, joint contractures, and the characteristic cauliflower ear and hitchhiker thumb.
- Normal
- Normal via SLC26A2 antiporter
- SLC26A2-Deficient
- Reduced (loss of function)
- Clinical Consequence
- Intracellular sulfate depletion
- Normal
- Normal chondroitin sulfate chains
- SLC26A2-Deficient
- Undersulfated chains
- Clinical Consequence
- Weak cartilage matrix
- Normal
- Ordered columnar arrangement
- SLC26A2-Deficient
- Disorganised, cystic
- Clinical Consequence
- Short, broad long bones
- Normal
- Full passive range
- SLC26A2-Deficient
- Multiple contractures and subluxations
- Clinical Consequence
- Stiff joints - hip, knee, elbow
Cartilage of the pinna depends on sulfated proteoglycans for its firm elastic scaffold. When SLC26A2 is defective, the pinna cartilage is structurally weak, the matrix cannot retain water, and the ear develops cystic swellings (often bilateral) in the first weeks of life. The cysts fibrose and calcify, leaving the classic "cauliflower" appearance - pathognomonic.
First metacarpal is short, broad, and proximally placed with a hypermobile metacarpophalangeal (MCP) joint. The thumb assumes a radial-abducted posture at rest - like a hitchhiker. Function is poor (cannot oppose), but the thumb is present (NOT absent - distinguishes from thrombocytopenia-absent radius).
Cervical kyphosis is mechanical and postural in most infants. With neck extension (positioning) and growth of the vertebral bodies, the kyphosis corrects. Only fixed, severe (greater than 60 degrees) kyphosis with cord signal change on MRI needs surgical fusion.
Classification and Types
SLC26A2 Phenotypic Series (Same Gene, Three Phenotypes)
- Severity
- Mildest - ambulant
- Key Features
- Hitchhiker thumb, cauliflower ear, clubfoot, normal IQ
- Lethality
- Viable - normal life expectancy
- Genotype
- Compound het or homozygous missense
- Severity
- Severe - non-lethal but fragile
- Key Features
- Severe micromelia, round face, cleft palate, hitchhiker toes
- Lethality
- Often neonatal lethal if respiratory failure
- Genotype
- Missense / splice - severe loss of function
- Severity
- Most severe
- Key Features
- Very short limbs, short thorax, soft skull
- Lethality
- Perinatally lethal
- Genotype
- Null mutations (frameshift, nonsense)
The three phenotypes reflect residual SLC26A2 function: missense variants preserve some function (diastrophic dysplasia), more severe variants cause atelosteogenesis, and null variants cause achondrogenesis 1B.
Clinical Assessment
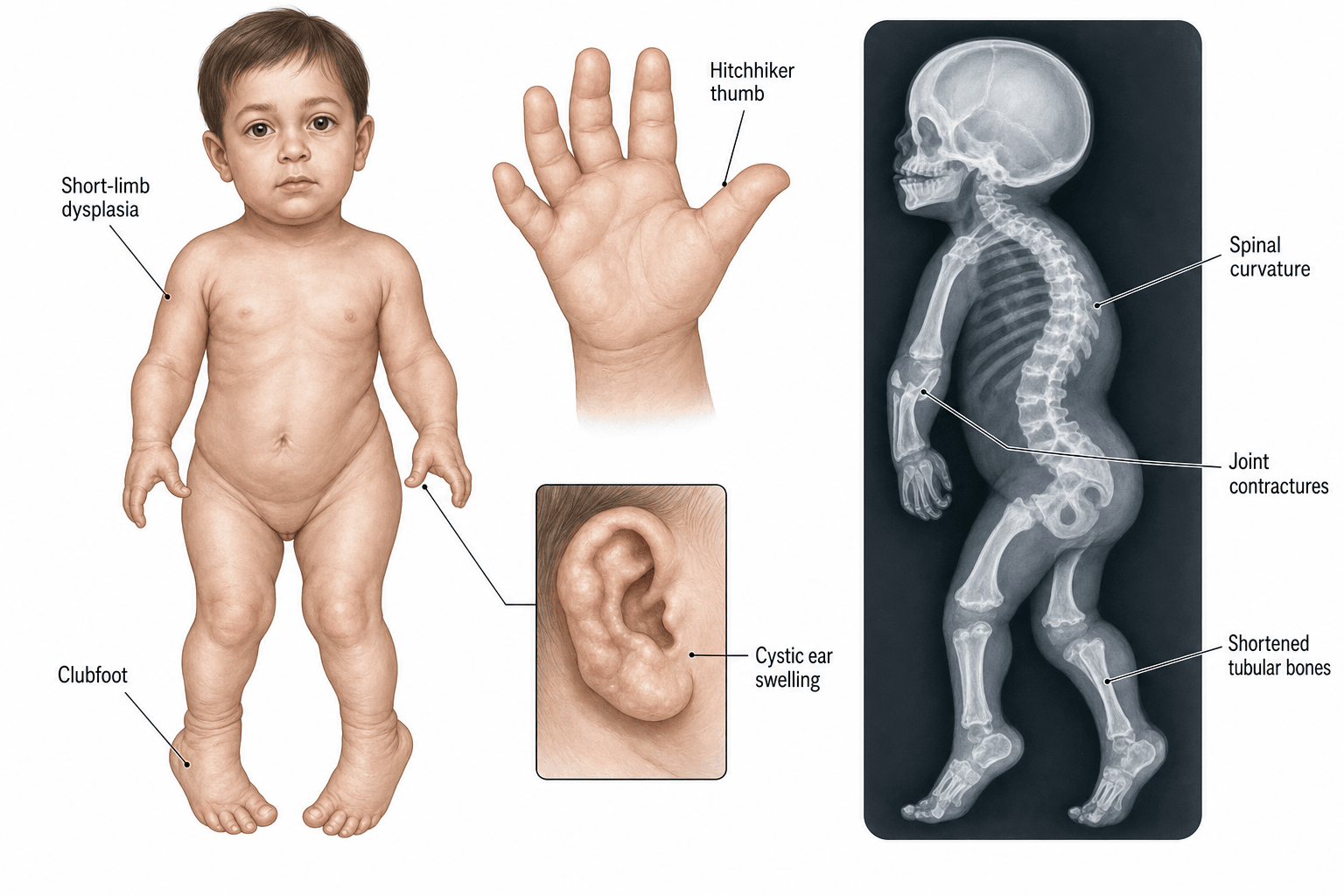
- Pregnancy: Polyhydramnios (impaired fetal swallowing from micrognathia), breech presentation
- Family: Consanguinity, prior affected sibling, ethnic background (Finnish, French-Canadian)
- Birth: Low birthweight, micromelia noted at delivery, cystic ear swelling in first weeks
- Development: Motor delay from joint stiffness, normal social and language milestones
- Limbs: Micromelia (rhizomelic and mesomelic), limited elbow extension, knee flexion contracture
- Hands: Hitchhiker thumb, brachydactyly, ulnar deviation, limited MCP motion
- Spine: Cervicothoracic kyphosis, scoliosis, lumbar hyperlordosis
- Feet: Rigid equinovarus adductus, hitchhiker great toe
- Face: Micrognathia, cleft palate (30 percent), normal facies otherwise
- Ears: Cystic pinnae (postnatal), then firm calcified cauliflower ear
Every newly diagnosed child needs:
- Lateral cervical X-ray (neutral) - measure C3-C5 kyphosis angle
- Flexion-extension lateral views - assess for instability
- MRI if kyphosis greater than 30 degrees - exclude cord compression
- Anaesthesia consult for future surgery - difficult intubation predicted
Red flags: Neurological deficit, progressive kyphosis, atlantoaxial instability on flexion-extension.
- Diastrophic Dysplasia
- Markedly reduced (35-45 cm)
- Distinguishing from Other Dysplasias
- Achondroplasia: short limbs only
- Diastrophic Dysplasia
- Present (pathognomonic)
- Distinguishing from Other Dysplasias
- Achondroplasia: trident, no hitchhiker
- Diastrophic Dysplasia
- Present (pathognomonic)
- Distinguishing from Other Dysplasias
- Achondroplasia: normal ears
- Diastrophic Dysplasia
- Short, broad, flared metaphyses
- Distinguishing from Other Dysplasias
- Achondroplasia: V-shaped epiphyses
- Diastrophic Dysplasia
- Kyphosis at C3-C5 common
- Distinguishing from Other Dysplasias
- Achondroplasia: foramen magnum stenosis
Achondroplasia has rhizomelic short limbs but trident hand, frontal bossing, normal ears, and NO hitchhiker thumb. Diastrophic dysplasia has micromelia with hitchhiker thumb, cauliflower ear, and rigid clubfeet. The two are easily separated clinically; genetic testing (FGFR3 vs SLC26A2) confirms.
Investigations
Imaging and Genetic Workup
Views: Babygram (whole-body AP), lateral skull, lateral cervical, AP chest, AP pelvis, AP long bones, AP hands and feet
Look for: Short first metacarpal, broad metaphyses, medial deviation of talus, cervical kyphosis, scoliosis
Views: Lateral neutral, flexion-extension lateral, MRI if kyphosis greater than 30 degrees
Look for: Cervicothoracic kyphosis angle, C1-C2 instability, cord compression
Test: SLC26A2 gene sequencing (Sanger or panel) - 95 percent sensitivity. If only one variant found, look for large deletions (MLPA).
Look for: Biallelic pathogenic variants (homozygous or compound heterozygous)
Views: Whole-spine EOS radiograph annually (low dose), MRI of spine at 3-5 years
Look for: Cobb angle progression, sagittal alignment, neural axis abnormalities
Indication: Major orthopaedic surgery (spine, hip, knee, foot)
CT: Bone detail for pedicle screw planning. MRI: Soft tissue and cartilage, neural elements
The first metacarpal is short and ovoid on plain radiograph - this is the radiographic equivalent of the hitchhiker thumb. Combined with the cauliflower ear, plain films alone are highly suggestive; genetic testing confirms the diagnosis and provides recurrence-risk information.
- Gene
- SLC26A2
- Hand
- Hitchhiker thumb
- Ear
- Cauliflower ear
- Spine
- Cervical kyphosis, scoliosis
- Gene
- FGFR3
- Hand
- Trident hand
- Ear
- Normal
- Spine
- Foramen magnum stenosis
- Gene
- FGFR3
- Hand
- Mild brachydactyly
- Ear
- Normal
- Spine
- Usually normal
- Gene
- SLC26A2 (allelic)
- Hand
- Hitchhiker (severe)
- Ear
- Cystic ear
- Spine
- Cervical instability, often lethal
- Gene
- FGFR3
- Hand
- Trident, short
- Ear
- Normal
- Spine
- Platyspondyly - lethal
DIASDiastrophic Dysplasia Diagnostic Tetrad
Hook:A newborn with DIAS has diastrophic dysplasia until proven otherwise!
Management Algorithm
Spine Management (Cervical and Thoracolumbar)
Goal: Prevent cord compression, halt progressive scoliosis, preserve function
Spine Treatment Protocol
Cervical kyphosis: Monitor with serial lateral X-rays every 3-6 months
Positioning: Prone or supine with neck in extension (roll under shoulders)
Indications for surgery: Progressive kyphosis greater than 60 degrees, neurological deficit, cord signal change on MRI
Surgery: Posterior cervical fusion (in-situ or with instrumentation) ± decompression
Scoliosis surveillance: Annual EOS radiograph from 2 years
Bracing: Cobb angle 20-40 degrees in growing child (limited efficacy in dystrophic curves)
Custom TLSO: Brace 18-23 hours per day until maturity or surgery
Surgery threshold: Curve 50+ degrees, progression greater than 10 degrees/year despite bracing
Surgery: Posterior spinal fusion with segmental instrumentation. Growing rods under 10 years if significant growth remains.
Pitfalls: High pseudarthrosis rate (small bones, soft bone) - maximise fixation density
Cervical kyphosis in diastrophic dysplasia is often self-correcting. Operate only for severe, progressive curves (greater than 60 degrees) with neurological involvement. For scoliosis, do not delay surgery until maturity - early fusion with modern segmental instrumentation gives the best long-term outcome and prevents restrictive lung disease.
Complications
- Incidence
- Less than 10 percent
- Risk Factors
- Kyphosis greater than 60 degrees, C1-C2 instability
- Management
- MRI surveillance, posterior fusion if progressive
- Incidence
- Greater than 50 percent by age 10
- Risk Factors
- Dystrophic curve pattern, early onset
- Management
- Brace 20-40 degrees, fusion 50+ degrees
- Incidence
- Common if severe scoliosis
- Risk Factors
- Early-onset severe thoracic curve
- Management
- Early fusion, pulmonary function tests
- Incidence
- Greater than 50 percent
- Risk Factors
- Capsular laxity, femoral deformity
- Management
- Open reduction ± osteotomy, THR later
- Incidence
- Greater than 60 percent
- Risk Factors
- Dysplastic tarsals, fibrous ligaments
- Management
- Serial releases, eventual talectomy
- Incidence
- Mixed, 30-50 percent
- Risk Factors
- Otitis media, ossicular abnormality
- Management
- Grommets, hearing aids, surveillance
- Incidence
- 10-20 percent after fusion
- Risk Factors
- Small pedicles, soft bone
- Management
- Maximise fixation, bone graft, brace
- Incidence
- Greater than 30 percent
- Risk Factors
- Micrognathia, short neck, cervical instability
- Management
- Awake fibreoptic intubation, cervical protection
High-risk for general anaesthesia: micrognathia, small mouth, short stiff neck with cervical kyphosis, atlantoaxial instability in some, restrictive lung disease from severe scoliosis. Pre-op: lateral cervical X-ray + flexion-extension views, MRI if instability suspected, awake fibreoptic intubation, experienced paediatric anaesthetist, spinal cord monitoring for spine surgery.
Outcomes and Prognosis
- Realistic Outcome
- Near normal life expectancy
- Limiting Factors
- Respiratory failure, anaesthesia complications
- Optimisation
- Early scoliosis management, careful airway
- Realistic Outcome
- Community ambulation in 80 percent
- Limiting Factors
- Knee contracture, hip dislocation, rigid clubfoot
- Optimisation
- Early standing, multi-level surgery, orthotics
- Realistic Outcome
- Independent ADL achievable in 70 percent
- Limiting Factors
- Hand function, joint stiffness
- Optimisation
- Hand therapy, tendon transfers, assistive devices
- Realistic Outcome
- Normal intelligence - mainstream education
- Limiting Factors
- School absence from surgery, accessibility
- Optimisation
- School support, career counselling
- Realistic Outcome
- 100-140 cm typical
- Limiting Factors
- Disproportionate short stature
- Optimisation
- Realistic expectation, growth hormone not effective
Best prognosis: Early multi-disciplinary care, compliant orthotic use, well-timed scoliosis surgery, hip and knee surgery before age 4-5. Poor prognosis: Severe scoliosis with cardiopulmonary compromise, untreated hip dislocation, fixed knee contracture, recurrent untreated clubfoot. Realistic expectations: Independent community ambulation achievable in 80 percent with comprehensive care. Final height 100-140 cm typical. Normal life expectancy.
The topic focuses on childhood surgery and mentions "THR later" only once, but - because life expectancy is normal - the adult sequelae are a genuine and examinable part of the disease:
- Premature osteoarthritis of the hips and knees is near-universal in adulthood, driven by the dysplastic, incongruent joints, residual contractures and altered mechanics. Disabling hip and knee pain in early-to-middle adult life is common and is often the reason an adult with diastrophic dysplasia presents to an arthroplasty surgeon.
- Arthroplasty is technically demanding in this short-limbed, dysplastic skeleton: the bones are small and deformed (frequently needing small, custom or paediatric implants, and sometimes custom femoral stems), there is often retained osteotomy hardware and altered proximal-femoral/condylar geometry from prior childhood surgery, fixed flexion contractures and soft-tissue tightness complicate balancing, and acetabular dysplasia/protrusio may require augmentation. Meticulous templating, the availability of small/custom implant sets, and planned soft-tissue releases are essential, and outcomes - while worthwhile for pain - are less predictable than in routine arthroplasty.
- Cervical-spine and airway risk persists into adulthood: the same difficult-airway and cervical-instability considerations from childhood apply to the adult coming for hip or knee replacement - obtain cervical imaging and plan the airway before any adult arthroplasty.
Exam point: diastrophic dysplasia is not only a paediatric problem - the adult develops early hip and knee OA and frequently needs arthroplasty with small/custom implants, careful management of prior deformity/hardware and contractures, and the same cervical-airway precautions as in childhood.
Guidelines, Registries & Global Practice
- Worldwide incidence approximately 1 in 100,000 live births - rare but pan-ethnic
- Finnish enrichment - 1 in 33,000 births in Finland due to founder mutation c.862C greater than T (p.Arg279Trp) on SLC26A2
- French-Canadian, African American, and Amish populations also have elevated carrier frequency
- Autosomal recessive - consanguinity in the family history is a major risk factor
- No sex predilection - equal incidence in males and females
- High-resource centres (Skeletal Dysplasia Registry, paediatric spine units): multi-disciplinary care with geneticist, paediatric orthopaedic surgeon, spine surgeon, anaesthetist, hand surgeon, physiotherapist, audiologist
- Limited-resource settings: Ponseti casting, sequential soft-tissue releases, and bracing can achieve plantigrade feet and ambulation in most children, even without access to advanced imaging or instrumentation
- Cervical kyphosis observation with serial X-rays is universally applicable; fusion reserved for severe progressive cases
- Genetic counselling is essential everywhere - diagnosis, recurrence risk, prenatal testing options
- Diagnosis emphasis
- Clinical features + SLC26A2 sequencing; proteoglycan sulfation biology
- Surgery thresholds
- Cervical fusion greater than 60 degrees with deficit; scoliosis fusion 50+ degrees
- Diagnosis emphasis
- Skeletal survey at birth; SLC26A2 panel for confirmation
- Surgery thresholds
- Foot: posteromedial release if Ponseti fails; hip: open reduction if DDH persists
- Diagnosis emphasis
- Multi-disciplinary team approach; genetic diagnosis; airway assessment
- Surgery thresholds
- Growing rods for very young with severe curves; definitive fusion for older children
- Diagnosis emphasis
- SLC26A2 sequencing and deletion analysis
- Surgery thresholds
- Centralised care in specialist dysplasias centres for complex surgery
There is no dedicated implant registry for diastrophic dysplasia (no arthroplasty is typical before skeletal maturity), but international rare-disease registries track the natural history, surgical outcomes, and genotype-phenotype correlations. The ERN-BOND and ISDS maintain consensus surveillance protocols and host multi-centre research.
Record in every newly diagnosed child with diastrophic dysplasia:
- SLC26A2 biallelic mutations confirmed (or sequencing pending)
- Cervical spine X-ray at presentation and every 3-6 months in infancy
- Whole-spine radiograph annually from 2 years of age
- Audiology assessment at 1 year; anaesthesia review before any surgery
- Genetic counselling completed - 25 percent recurrence risk
A missed cervical cord compression or a poorly planned anaesthesia can be fatal. Always document the cervical spine status and have a difficult-airway plan.
Controversies & Areas of Uncertainty
The 1991 Poussa series suggested many patients needed cervical fusion. The 2001 Remes revisit showed most resolve spontaneously. Current consensus: observe most cases with serial imaging; operate only on curves greater than 60 degrees, progressive curves, or those with neurological involvement. Long-term outcome data remain limited.
Bracing is largely ineffective in dystrophic curves. Growing rods are an option for under-8-year-olds with severe curves, but multiple lengthenings carry morbidity. Definitive posterior fusion is now performed at younger ages with modern segmental instrumentation, but the high pseudarthrosis rate (10-20 percent) and the impact on remaining growth are debated.
Early extensive posteromedial release maximises plantigrade alignment but recurrence is the rule and re-do surgery is common. Staged, less aggressive releases preserve more joint motion but require more procedures. There is no consensus on the optimal strategy - decision is surgeon- and family-preference driven.
Distraction osteogenesis can add 15-25 cm to femur and tibia. However, the high complication rate (joint stiffness, regenerate fracture, deformity, pin-site infection, prolonged rehab) and the risk to existing ambulatory function make most centres cautious. Decision is highly individualised.
MCQ Practice Points
Q: What gene is mutated in diastrophic dysplasia, and what is the inheritance pattern? A: SLC26A2 (originally called DTDST) on chromosome 5q32-q33.1, encoding a sulfate-chloride antiporter. Autosomal recessive - both parents are carriers, with 25 percent recurrence risk. Mutations in the same gene cause atelosteogenesis type II and achondrogenesis type 1B (allelic series).
Q: What are the two most pathognomonic clinical signs of diastrophic dysplasia in a newborn? A: Cystic swelling of the pinnae (cauliflower ear) and hitchhiker thumb. The cauliflower ear appears in the first weeks of life and is essentially diagnostic. The hitchhiker thumb is a radially abducted, proximally placed thumb with hypermobile MCP - so named because it resembles a hitchhiker's thumb. Both are present in over 80 percent of affected infants.
Q: A 2-year-old with diastrophic dysplasia has rigid bilateral equinovarus despite 5 Ponseti casts. What is the next step? A: Modified Ponseti is unlikely to fully correct clubfoot in diastrophic dysplasia. The dysplasia is in the bones and cartilages themselves, not just the soft tissues. After partial correction with casting, the standard next step is posteromedial soft-tissue release (Cincinnati or Turco incision) at age 1-4 years. Recurrence is common (greater than 60 percent) and multiple releases are often required.
Q: How do you distinguish diastrophic dysplasia from achondroplasia clinically? A: Both cause short-limbed dwarfism, but achondroplasia has trident hand, frontal bossing, normal ears, foramen magnum stenosis, and rhizomelic limb shortening. Diastrophic dysplasia has hitchhiker thumb, cauliflower ear, micromelia, cervical kyphosis, and rigid clubfoot. Genetic testing (FGFR3 vs SLC26A2) confirms.
Q: Why is general anaesthesia particularly hazardous in a patient with diastrophic dysplasia? A: The combination of micrognathia, small mouth opening, short stiff neck, and potential cervical instability makes intubation very difficult. Awake fibreoptic intubation is the technique of choice, and an experienced paediatric anaesthetist should be present. Restrictive lung disease from severe scoliosis compounds the respiratory risk.
Exam Viva Scenarios
Practise clinical reasoning and management decisions out loud
“A 35-week preterm infant is born to consanguineous parents of Somali origin. The neonate has severe short-limbed dwarfism, bilateral cystic swellings of both pinnae, radially abducted thumbs (hitchhiker posture), and rigid bilateral equinovarus feet. There is a small midline cleft of the soft palate. The cervical spine X-ray shows 35 degrees of kyphosis at C3-C5. What is the most likely diagnosis, what genetic test confirms it, and what is the immediate orthopaedic plan?”
“A 6-year-old girl with genetically confirmed diastrophic dysplasia presents with a 12-month history of progressive scoliosis. Her whole-spine EOS radiograph shows a 55-degree right thoracic curve (T5-T11) with 35 degrees of thoracolumbar kyphosis and 50 degrees of lumbar hyperlordosis. She has had a single previous general anaesthetic for cleft palate repair without complication. MRI shows no intraspinal abnormality. She is community-ambulant but struggles with stairs. What is your management plan?”
Diagnosis at Birth
- SLC26A2 (5q32-q33.1) - sulfate-chloride antiporter, autosomal recessive
- Hitchhiker thumb (radial abduction, hypermobile MCP) - pathognomonic
- Cauliflower ear (cystic pinnae in first weeks) - pathognomonic
- Rigid bilateral equinovarus - resistant to Ponseti
- Micromelia (rhizomelic and mesomelic), short first metacarpal
Genetics and Phenotype
- Same gene causes atelosteogenesis type II and achondrogenesis 1B (allelic series)
- Missense - partial function - diastrophic dysplasia
- Severe loss of function - atelosteogenesis type II
- Null mutations - achondrogenesis 1B (lethal)
Spine Management
- Cervical kyphosis C3-C5: most resolve spontaneously - serial X-rays
- Operate cervical kyphosis if greater than 60 degrees and progressive
- Scoliosis: brace 20-40 degrees, fusion 50+ degrees - dystrophic curves do not brace well
- Intra-op neuromonitoring mandatory - small cord, high pseudarthrosis rate
Hip and Knee
- Hip dysplasia in greater than 50 percent - Pavlik if reducible, open reduction if persistent
- Knee flexion contracture greater than 30 degrees prevents standing
- Distal femoral extension osteotomy for fixed knee deformity
- Operate knee and hip before age 4-5 to enable ambulation
Foot and Hand
- Clubfoot: modified Ponseti for partial correction, posteromedial release in 80 percent
- Recurrence is the norm - multiple releases, eventual talectomy or triple arthrodesis
- Hitchhiker thumb: first web space deepening (4-flap Z-plasty) at 1-4 years
- Opponensplasty (abductor digiti minimi or FDS) for opposition
Anaesthesia and Airway
- Difficult intubation predicted - micrognathia, short neck, cervical instability
- Pre-op: lateral cervical X-ray + flexion-extension views
- Awake fibreoptic intubation is technique of choice
- Spinal cord monitoring for spine surgery
Surveillance Schedule
- Cervical X-ray every 3-6 months in infancy, annually thereafter
- Whole-spine EOS radiograph from 2 years, then annually
- Hip ultrasound in newborn, then clinical + X-ray surveillance
- Audiology at 1 year; genetic counselling by 1 year (25 percent recurrence risk)
Outcomes
- Community ambulation in 80 percent with comprehensive care
- Final adult height 100-140 cm (typical short-limbed dwarfism)
- Normal intelligence - mainstream education achievable
- Realistic goal: independent ADL, plantigrade feet, corrected spine, functional hands
Evidence Base and Key Trials
The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping
- Positional cloning identified SLC26A2 (originally DTDST) on chromosome 5q32-q33.1 as the gene responsible for diastrophic dysplasia using fine-structure linkage disequilibrium in the genetically isolated Finnish population
- The encoded protein is a transmembrane sulfate-chloride antiporter - the first skeletal dysplasia shown to be a disorder of sulfate metabolism
- Homozygous and compound heterozygous loss-of-function mutations correlate with the diastrophic dysplasia phenotype, opening a new field in skeletal biology
Atelosteogenesis type II is caused by mutations in the diastrophic dysplasia sulfate-transporter gene (DTDST): evidence for a phenotypic series involving three chondrodysplasias
- Mutations in the same SLC26A2 gene that cause diastrophic dysplasia also cause atelosteogenesis type II and (with achondrogenesis 1B) form a phenotypic series
- Genotype-phenotype correlation: missense variants that preserve partial sulfate transport produce diastrophic dysplasia, while more severe loss-of-function variants produce atelosteogenesis type II
- The phenotypic series provides the framework for understanding the clinical spectrum of skeletal dysplasias caused by the same gene
The spine in diastrophic dysplasia
- Systematic radiographic survey of 50 Finnish patients with diastrophic dysplasia showed cervical kyphosis at C3-C5 in a third of newborns and progressive scoliosis in more than half by skeletal maturity
- Cervical kyphosis often resolves with growth and neck extension - surgical fusion is reserved for severe, progressive curves with neurological involvement
- Scoliosis typically begins in early childhood, becomes severe through the growth spurt, and frequently requires long-segment posterior fusion
Cervical kyphosis in diastrophic dysplasia
- Long-term radiographic follow-up of 88 Finnish patients with diastrophic dysplasia showed that cervical kyphosis at C3-C5 present in infancy improves spontaneously with growth in the majority of cases
- Severe, persistent kyphosis (greater than 60 degrees) and C1-C2 instability are the principal indications for posterior cervical fusion
- Careful neurological examination and serial imaging identify the small subgroup who need surgery; the majority can be managed non-operatively
Growth in diastrophic dysplasia
- Longitudinal growth study of 121 Finnish patients defined the natural history of stature in diastrophic dysplasia from birth to skeletal maturity
- Mean birth length is markedly reduced and the growth curve remains well below the 3rd centile throughout childhood and adolescence
- Mean final adult height is approximately 110-140 cm in females and 130-150 cm in males - a typical short-limbed adult stature