COMP / COL9A1 / COL9A2 / COL9A3 Mutations | Small Irregular Epiphyses | Early OA | Spine Spared
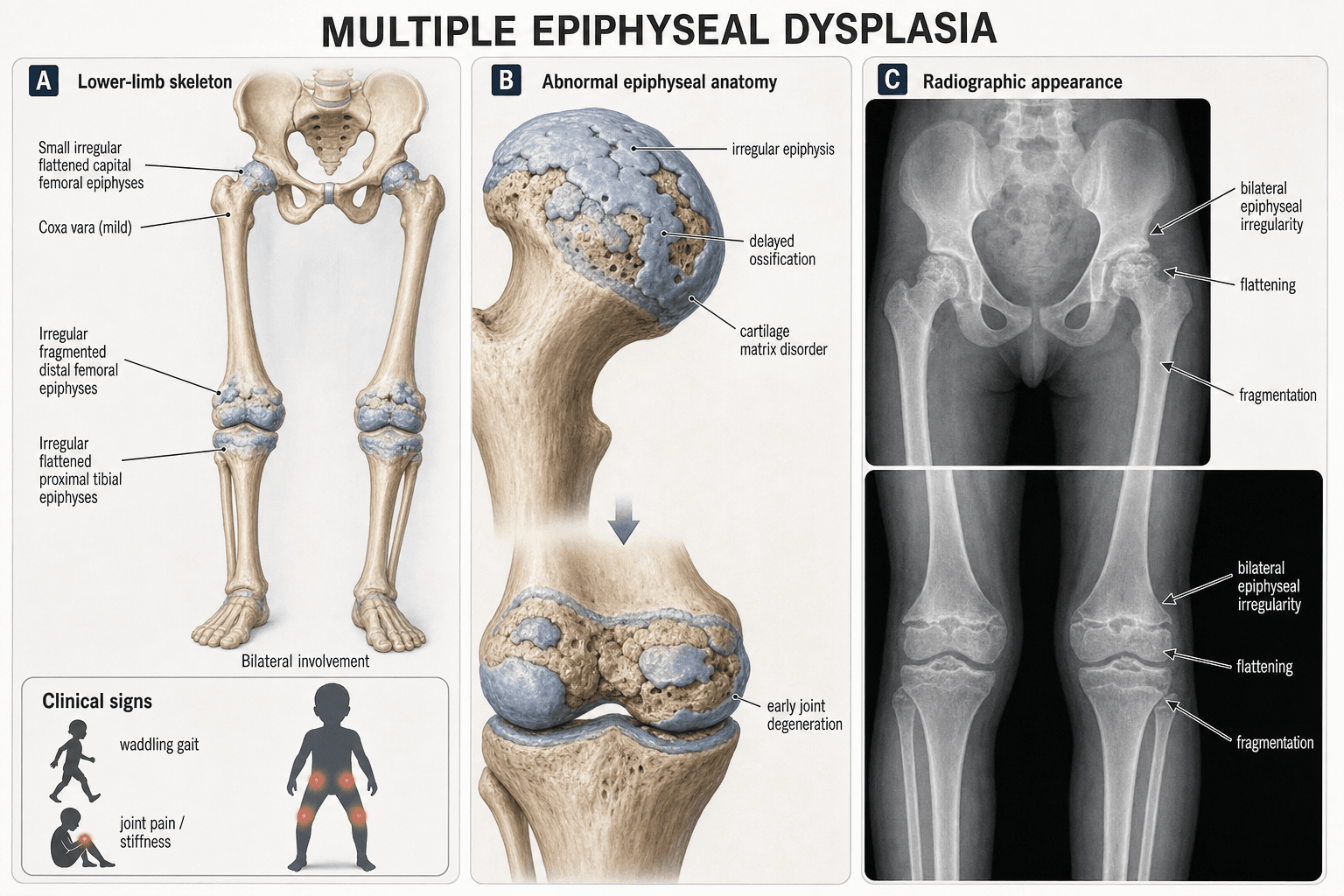
- Small, irregular, delayed epiphyses at multiple joints (hips, knees, ankles) with spine largely spared
- Waddling gait and joint pain in early childhood — commonly misdiagnosed as bilateral Perthes
- COMP mutations (most common, approximately 50 percent) affect cartilage extracellular matrix assembly
- Early-onset osteoarthritis is the dominant long-term morbidity — often needing THA or TKA by age 40-50
- AP pelvis X-ray: small, fragmented femoral heads with preserved acetabular coverage (unlike Perthes)
- “MED spines are normal or near-normal — if significant platyspondyly, think spondyloepiphyseal dysplasia
- “Bilateral symmetric epiphyseal involvement distinguishes MED from Perthes (sequential unilateral)
- “Waddling gait + short stature + stiff joints + normal spine = MED until proven otherwise
- “Ask about family history — autosomal dominant inheritance is most common
Most common: COMP mutation (AD). Also COL9A1, COL9A2, COL9A3 (AD or AR), MATN3 (AD). All affect cartilage extracellular matrix. Penetrance is variable; some carriers are asymptomatic.
Short stature + waddling gait + joint stiffness/pain. Onset typically 2-10 years. Stature usually 2-3 SD below mean. Spine is characteristically spared (no significant platyspondyly).
Small, irregular, delayed epiphyses at multiple joints bilaterally. Hips, knees, ankles, shoulders affected. Femoral heads are small and fragmented but acetabular coverage is preserved (unlike Perthes). Epiphyses ossify late.
MED is symmetric, familial, and spares the acetabulum. Perthes is sequential (one side then the other), not familial, and shows acetabular changes. In MED, both femoral heads are small at the same time; in bilateral Perthes, they are at different stages.
- MED
- AD (usually), positive family history
- Bilateral Perthes
- Sporadic, no family history
- Key Discriminator
- Ask about affected relatives
- MED
- Both hips symmetric and simultaneous
- Bilateral Perthes
- Sequential — second hip 1-3 years after first
- Key Discriminator
- Symmetry favors MED
- MED
- Normal coverage, no acetabular changes
- Bilateral Perthes
- Acetabular dysplasia and changes develop
- Key Discriminator
- Acetabular involvement = Perthes
- MED
- Knees, ankles, shoulders also affected
- Bilateral Perthes
- Isolated to hips
- Key Discriminator
- Multi-joint = MED
- MED
- Normal or minimal change
- Bilateral Perthes
- Normal
- Key Discriminator
- Significant platyspondyly = neither (think SED)
SPAREDMED vs Perthes vs SED
Hook:The SPINE is SPARED in MED — if the spine is abnormal, think SED not MED!
Overview and Epidemiology
Multiple Epiphyseal Dysplasia is one of the most common skeletal dysplasias and a leading genetic cause of early-onset osteoarthritis. It is frequently misdiagnosed as bilateral Perthes disease, leading to inappropriate treatment. Recognizing MED early allows genetic counselling, avoids unnecessary surgery in childhood, and sets realistic expectations for the likely need for joint replacement in early adulthood.
- Prevalence: Approximately 1 in 10,000 live births
- Inheritance: Predominantly autosomal dominant; rare AR forms
- Sex: Equal male-to-female ratio (AD forms)
- Geography: Worldwide distribution, no ethnic predilection
- Short stature: Mild, usually 2-3 SD below mean (not as severe as achondroplasia)
- Early OA: Most patients develop significant OA by the third to fourth decade
- Functional limitation: Progressive pain and stiffness in hips, knees, ankles
- Joint replacement: Often required by age 30-50 years
Pathophysiology
Normal articular cartilage relies on the coordinated function of collagen II (structural framework), collagen IX (cross-linking and fibril stability), and COMP (calcium-binding protein that facilitates collagen fibrillogenesis). In MED, mutations in COMP, COL9A1-3, or MATN3 disrupt the cartilage extracellular matrix assembly. COMP mutations cause protein misfolding and retention in the rough endoplasmic reticulum of chondrocytes, triggering ER stress and premature chondrocyte apoptosis. COL9 mutations weaken collagen fibril cross-linking, reducing the tensile strength of the cartilage matrix. The net result is defective endochondral ossification — epiphyses ossify late and irregularly, producing small, misshapen articular surfaces that are biomechanically inferior and predispose to early osteoarthritis.
- Protein
- Cartilage oligomeric matrix protein
- Inheritance
- AD
- Frequency
- Approximately 50 percent of cases
- Phenotype Severity
- Moderate to severe
- Protein
- Collagen IX alpha-2
- Inheritance
- AD
- Frequency
- Approximately 15-20 percent
- Phenotype Severity
- Mild to moderate
- Protein
- Collagen IX alpha-3
- Inheritance
- AD
- Frequency
- Approximately 10-15 percent
- Phenotype Severity
- Mild to moderate
- Protein
- Collagen IX alpha-1
- Inheritance
- AD or AR
- Frequency
- Rare (less than 5 percent)
- Phenotype Severity
- Variable, AR form more severe
- Protein
- Matrilin-3
- Inheritance
- AD
- Frequency
- Approximately 10 percent
- Phenotype Severity
- Milder phenotype
Normal COMP: Secreted calcium-binding protein, facilitates collagen II and IX fibrillogenesis
Mutated COMP: Misfolds in the rough ER of chondrocytes
Cellular consequence: ER stress response, premature chondrocyte apoptosis
Tissue result: Defective endochondral ossification, small irregular epiphyses
Clinical: Similar mechanism to pseudoachondroplasia (also COMP, but more severe phenotype)
Normal collagen IX: Heterotrimer of alpha-1, alpha-2, alpha-3 chains; cross-links collagen II fibrils
Mutated COL9: Disrupted heterotrimer assembly or fibril cross-linking
Tissue result: Weak cartilage matrix with reduced tensile strength
Clinical: Tends to be milder than COMP-MED, but still causes early OA
Note: COL9A1 AR mutations cause a more severe phenotype with myopathy
COMP-COLGenetic Causes of MED
Hook:COMP-COL: COMP and COLlagen IX are the two main genetic pathways in MED!
Classification and Types
Genetic Classification of MED
- Gene
- COMP
- Key Pathology
- ER stress, chondrocyte apoptosis
- Severity
- Moderate-severe
- Distinctive Feature
- Most common; overlaps pseudoachondroplasia spectrum
- Gene
- COL9A2
- Key Pathology
- Collagen IX cross-link failure
- Severity
- Mild-moderate
- Distinctive Feature
- AD; common in Northern European populations
- Gene
- COL9A3
- Key Pathology
- Collagen IX cross-link failure
- Severity
- Mild-moderate
- Distinctive Feature
- AD; clinically similar to EDM2
- Gene
- SLC26A2 (DTDST)
- Key Pathology
- Sulfate transporter defect
- Severity
- Variable
- Distinctive Feature
- AR; overlaps with diastrophic dysplasia spectrum
- Gene
- MATN3
- Key Pathology
- Matrilin-3 extracellular matrix defect
- Severity
- Mild
- Distinctive Feature
- AD; relatively mild OA phenotype
- Gene
- COL9A1
- Key Pathology
- Collagen IX alpha-1 chain defect
- Severity
- Variable
- Distinctive Feature
- AD or AR; AR form has myopathy
Genetic classification guides prognosis and genetic counselling but does not currently change orthopaedic management, which remains supportive.
Long before genetic subtyping, MED was split clinically into two eponymous phenotypes that examiners still use:
- Fairbank type — the more severe, generalised form: many joints involved (hips, knees, ankles, shoulders and hands), more pronounced short stature, and small epiphyses throughout the skeleton.
- Ribbing type — the milder form: predominantly the hips, near-normal stature, often presenting only in adolescence or adulthood as premature hip osteoarthritis.
The split is descriptive and the two overlap with genotype (COMP tends toward the severe Fairbank pattern; the milder COL9 and MATN3 forms toward Ribbing), but it remains a useful clinical shorthand and a frequent viva term. Both share the cardinal MED features — bilateral symmetric epiphyseal dysplasia with a spared spine.
Clinical Assessment
- Onset: Age 2-10 years with waddling gait or joint pain
- Family history: Often positive (AD) — ask about relatives with early arthritis or joint replacement
- Gait: Waddling, Trendelenburg pattern from hip involvement
- Pain: Progressive joint pain, worse with activity, improving with rest
- Stiffness: Morning stiffness in affected joints, improves with movement
- Sports: Difficulty keeping up with peers, early fatigue
- Stature: Mild short stature (2-3 SD below mean, proportionate)
- Gait: Waddling, antalgic, or Trendelenburg
- Hips: Restricted IR, flexion and abduction; pain on log roll
- Knees: Varus or valgus deformity, restricted flexion, crepitus
- Ankles: Stiff subtalar joint, possible equinus
- Hands: Short, stubby fingers (brachydactyly) in some subtypes
- Spine: Essentially normal — no significant kyphosis or scoliosis
MED: Bilateral symmetric epiphyseal changes, positive family history (AD), normal acetabulum, multiple joints affected, spine spared, onset typically age 2-10
Bilateral Perthes: Sequential involvement (stages differ between sides), sporadic (no family history), acetabular changes develop, isolated to hips, onset typically age 4-8
Critical test: Look at the knees and ankles — if epiphyses at these joints are also small and irregular, the diagnosis is MED, not Perthes. Bilateral Perthes does not affect the knees or shoulders.
- Inheritance
- AD (usually)
- Spine
- Normal
- Other Joints
- Knees, ankles, shoulders
- Key Discriminator
- Small irregular epiphyses, symmetric, spared acetabulum
- Inheritance
- Sporadic
- Spine
- Normal
- Other Joints
- Hips only
- Key Discriminator
- Sequential stages, acetabular changes, no family history
- Inheritance
- AD (SED congenita) or X-linked
- Spine
- Platyspondyly, odontoid hypoplasia
- Other Joints
- Hips, knees
- Key Discriminator
- Significant spine involvement is the key differentiator
- Inheritance
- AD (COMP)
- Spine
- Mild platyspondyly
- Other Joints
- All epiphyses
- Key Discriminator
- More severe short stature, same COMP gene as MED
- Inheritance
- Sporadic / autoimmune
- Spine
- Delayed bone age, no platyspondyly
- Other Joints
- Universal delay
- Key Discriminator
- Epiphyseal dysgenesis, systemic features, TSH elevated
- Inheritance
- AR
- Spine
- Thoracolumbar gibbus, ovoid vertebrae
- Other Joints
- Dysostosis multiplex
- Key Discriminator
- Coarse facies, organomegaly, urinary GAGs
Many MED patients present with hip pain and bilateral femoral head abnormalities on X-ray. The default registrar diagnosis is bilateral Perthes. Always look at the knee X-rays before committing to this diagnosis — if the distal femoral and proximal tibial epiphyses are also small and irregular, you are dealing with MED. This distinction matters because Perthes is treated with containment (bracing, osteotomy) while MED requires supportive care and genetic counselling.
EPiphysesMED Clinical Features
Hook:Remember EPiphyses — the epiphyses are the problem in MED, and they are small, symmetric, and spare the spine!
Investigations
Diagnostic Workup
Views: AP pelvis, AP and lateral of both knees, AP and lateral ankles, lateral thoracolumbar spine, PA chest, AP and lateral of wrists (bone age)
Look for: Small, irregular, delayed epiphyses at multiple joints; preserved acetabular coverage; normal vertebral body height; delayed bone age; short metacarpals and phalanges in some subtypes
Clinical correlation: The pattern of epiphyseal involvement across multiple joints is diagnostic
Indication: Confirmed or suspected MED on clinical/radiographic grounds
Genes: COMP, COL9A1, COL9A2, COL9A3, MATN3, SLC26A2
Method: Targeted gene panel or whole exome sequencing
Yield: Approximately 50-60 percent of clinically diagnosed MED cases have an identifiable mutation in one of these genes
Indication: Assess articular cartilage integrity in symptomatic joints; pre-operative planning for joint preservation surgery
Findings: Thinning or loss of articular cartilage, subchondral cysts, osteophytes, marrow edema in early OA
Role: Primarily for surgical planning rather than diagnosis
Purpose: Exclude metabolic mimics (hypothyroidism, skeletal dysplasia screen)
Tests: TSH, free T4, calcium, phosphate, alkaline phosphatase, vitamin D
Note: Blood tests are normal in MED — they are performed to exclude metabolic and endocrine causes of epiphyseal dysplasia
The skeletal survey is the cornerstone of diagnosis. The key radiographic features are: (1) small, irregular epiphyses at multiple joints, (2) bilateral and symmetric involvement, (3) preserved acetabular depth and coverage, (4) normal or near-normal spine, and (5) delayed bone age. Genetic testing confirms the specific mutation but clinical management is determined by the phenotype, not the genotype.
A double-layered patella — a horizontal lucent cleft dividing the patella into anterior and posterior ossified layers on the lateral knee radiograph — is a rare but near-specific sign of recessive MED caused by SLC26A2 (DTDST) mutations (rMED / EDM4). It reflects the abnormal endochondral ossification of the patellar cartilage. Recessive MED also classically pairs the epiphyseal dysplasia with other DTDST-spectrum features — clubfoot, cleft palate, brachydactyly, and ear swelling or deformity. Spotting a double-layered patella on a knee film in a short-statured child with epiphyseal changes is a high-yield clue that points specifically to the recessive SLC26A2 form rather than the dominant COMP/COL9 types.

Management Algorithm
Childhood Management (Age 2-16)
Goal: Maintain mobility, manage pain, monitor epiphyseal development, provide genetic counselling
Childhood Treatment Protocol
Multidisciplinary team: Paediatric orthopaedics, genetics, physiotherapy
Genetic counselling: Discuss inheritance pattern, variable penetrance, implications for family planning
Activity modification: Avoid high-impact sports; encourage swimming and cycling
Physiotherapy: Maintain range of motion, strengthen periarticular muscles, gait training
Clinical monitoring: Height, gait, joint range of motion, pain assessment
Radiographic surveillance: Annual AP pelvis and knee X-rays to track epiphyseal development
Orthotics: Shoe modifications or AFOs for ankle involvement or leg-length discrepancy
Pain management: Activity modification, NSAIDs as needed, weight optimization
Osteotomy: Corrective femoral or tibial osteotomy for significant angular deformity (varus or valgus greater than 15 degrees) to improve joint biomechanics
Hip arthroscopy: Limited role; may be considered for labral tears or impingement in adolescents
Epiphysiodesis: Rarely indicated for leg-length discrepancy (usually mild in MED)
In childhood, the mainstay of treatment is non-operative. Avoid aggressive surgical interventions (such as femoral osteotomies for "containment" as in Perthes) because the epiphyseal pathology is genetic and systemic, not a vascular insult. Surgery is reserved for significant angular deformity or mechanical axis deviation. Genetic counselling is essential at diagnosis.
Complications
- Incidence / Timeline
- Near universal by the third to fourth decade
- Risk Factors
- All MED genotypes; severity varies
- Management
- Conservative then arthroplasty when quality of life impaired
- Incidence / Timeline
- Common in childhood and adolescence
- Risk Factors
- Asymmetric epiphyseal growth
- Management
- Corrective osteotomy if deformity greater than 15 degrees and symptomatic
- Incidence / Timeline
- Mild, usually less than 2 cm
- Risk Factors
- Asymmetric epiphyseal involvement
- Management
- Shoe raise, rarely epiphysiodesis
- Incidence / Timeline
- Higher than age-matched OA due to young age and bone quality
- Risk Factors
- Young age at arthroplasty, activity level
- Management
- Use durable bearings, plan for revision, activity modification
- Incidence / Timeline
- Significant — chronic pain, short stature, multiple surgeries
- Risk Factors
- Young age of onset, progressive disability
- Management
- Psychology referral, peer support, genetic counselling
The defining complication of MED is early-onset osteoarthritis. Unlike Perthes, where the goal is to preserve the femoral head shape through containment, in MED the cartilage matrix is inherently defective. No treatment can prevent the progression to OA. The role of the orthopaedic surgeon is to optimize joint biomechanics (correct deformity), maximize non-operative management, and time joint replacement appropriately — balancing the patient's pain and disability against the finite lifespan of a prosthesis in a young active patient.
Outcomes and Prognosis
- Typical OA Onset
- Late 20s to mid-30s
- Age at First Arthroplasty
- Mid-30s to mid-40s
- Long-term Function
- Good with arthroplasty; multiple revisions may be needed
- Typical OA Onset
- 30s to early 40s
- Age at First Arthroplasty
- 40s to 50s
- Long-term Function
- Favourable; later onset allows longer conservative management
- Typical OA Onset
- Late 30s to 40s
- Age at First Arthroplasty
- Late 40s to 50s
- Long-term Function
- Best prognosis among MED subtypes
- Typical OA Onset
- 20s (more severe)
- Age at First Arthroplasty
- 30s
- Long-term Function
- More guarded; earlier and more severe joint disease
Best prognosis: MATN3 and COL9 mutations, milder epiphyseal irregularity, compliant with activity modification, healthy BMI
Poorer prognosis: COMP mutations with severe epiphyseal irregularity, high BMI (accelerates OA), high-impact activity, early onset of symptoms (before age 5)
Key message for patients: MED does not affect life expectancy, intelligence, or systemic health. The morbidity is limited to the musculoskeletal system. With modern joint replacement, most patients achieve good functional outcomes and independent living.
Guidelines, Registries & Global Practice
- Worldwide prevalence: Approximately 1 in 10,000; no ethnic or geographic predilection
- AD forms: Most common globally; variable penetrance means mildly affected parents may be undiagnosed
- AR forms: Higher proportion in populations with consanguineous marriages (Middle East, South Asia)
- Underdiagnosis: Mild cases may not present until early adult OA, when the underlying dysplasia is not recognized
- High-resource: Genetic panel testing, paediatric orthopaedic multidisciplinary team, early THA with ceramic bearings
- Limited-resource: Diagnosis often made clinically and radiographically; genetic testing may be unavailable; arthroplasty options limited by implant cost and surgical expertise
- Universal principle: The diagnosis can be made on plain X-rays and clinical history alone; genetic testing confirms but is not required for management
- Physiotherapy and activity modification: Effective and low-cost across all resource settings
- Diagnosis Emphasis
- Clinical and radiographic diagnosis confirmed by genetic testing; skeletal survey essential
- Childhood Management
- Non-operative; activity modification, physiotherapy; avoid Perthes-type containment
- Adult / Arthroplasty
- Joint replacement when quality of life impaired; genetic counselling for family planning
- Diagnosis Emphasis
- Skeletal survey and genetic panel; differentiate from bilateral Perthes and SED
- Childhood Management
- Observation and supportive care; osteotomy only for significant angular deformity
- Adult / Arthroplasty
- THA/TKA with durable bearings; counsel on young-age revision risk
- Diagnosis Emphasis
- Clinical-radiographic diagnosis; genetics referral for confirmation and counselling
- Childhood Management
- Physiotherapy-led management; MDT approach with genetics and paediatric orthopaedics
- Adult / Arthroplasty
- Timed arthroplasty; consider lifelong orthopaedic follow-up for dysplasia patients
- Diagnosis Emphasis
- Focus on biomechanical assessment of angular deformity and joint congruity
- Childhood Management
- Corrective osteotomy principles when deformity alters mechanical axis significantly
- Adult / Arthroplasty
- Standard arthroplasty principles with attention to altered anatomy and bone quality
There is no dedicated arthroplasty registry for skeletal dysplasia patients. Most outcome data comes from small single-centre retrospective series. National joint registries (NJR, AOANJRR, AJRR) do not separately track MED patients, so survivorship data for THA/TKA in this population is derived from published case series rather than large-scale registry analysis. The evidence base is level 4 at best — observational and retrospective.
Every MED patient and family should receive genetic counselling covering:
- Inheritance pattern (AD in most cases — 50 percent transmission risk to offspring)
- Variable penetrance and expressivity (mildly affected parent may have severely affected child, and vice versa)
- Availability of prenatal or pre-implantation genetic diagnosis for family planning
- Psychological support for the child and family — chronic progressive condition with multiple surgeries
A missed genetic diagnosis can result in inappropriate treatment (Perthes containment for MED) and failure to counsel the family about reproductive risks.
Controversies & Areas of Uncertainty
Some surgeons advocate corrective osteotomy to improve joint biomechanics in childhood, while others argue that the genetic cartilage defect makes any mechanical correction futile in preventing OA. Current evidence is insufficient to strongly recommend for or against prophylactic osteotomy in MED.
Mesenchymal stem cell injections, PRP, and growth-factor therapies are being explored for cartilage preservation in skeletal dysplasias. No evidence supports disease modification in MED. These remain experimental and should not be offered outside clinical trials.
The optimal timing of THA/TKA in MED is debated: operate too early and face higher revision burden; delay too long and the patient suffers years of pain and functional decline. Most surgeons now favour operating when quality of life is significantly impaired, rather than waiting for an arbitrary age threshold.
Ceramic-on-ceramic, ceramic-on-XLPE, and metal-on-metal bearings each have proponents. No RCTs exist specific to the MED population. Current consensus favours ceramic-on-ceramic or ceramic-on-XLPE for young active patients, but the choice is individualized based on patient factors and surgeon experience.
MCQ Practice Points
Q: A 7-year-old presents with waddling gait. X-rays show bilateral small, irregular femoral heads, small distal femoral epiphyses, and a normal spine. What is the most likely diagnosis? A: Multiple Epiphyseal Dysplasia. The combination of bilateral symmetric epiphyseal irregularities at multiple joints (hips and knees) with a normal spine is characteristic. Bilateral Perthes would affect only the hips with sequential staging. Spondyloepiphyseal dysplasia would show platyspondyly.
Q: What is the most commonly mutated gene in Multiple Epiphyseal Dysplasia, and what is its inheritance pattern? A: COMP (cartilage oligomeric matrix protein), inherited in an autosomal dominant pattern. COMP mutations account for approximately 50 percent of MED cases. The gene is located on chromosome 19p13.1.
Q: How do you distinguish MED from bilateral Perthes disease on imaging? A: MED shows bilateral symmetric epiphyseal changes, preserved acetabular coverage, and involvement of other joints (knees, ankles, shoulders). Bilateral Perthes shows sequential involvement (different stages on each side), acetabular changes, and isolated hip involvement. A skeletal survey is the critical investigation.
Q: What is the pathogenesis of epiphyseal abnormalities in COMP-related MED? A: COMP mutations cause protein misfolding, leading to retention in the rough endoplasmic reticulum of chondrocytes. This triggers ER stress and premature chondrocyte apoptosis, disrupting endochondral ossification. The result is delayed and irregular epiphyseal ossification, producing small, misshapen articular surfaces prone to early osteoarthritis.
Q: What is the appropriate management of a 5-year-old with newly diagnosed MED and bilateral hip pain? A: Non-operative management: activity modification (swimming, cycling; avoid high-impact sports), physiotherapy for range of motion and gait, NSAIDs as needed for pain, and annual clinical and radiographic monitoring. Do not treat like Perthes — containment bracing and femoral osteotomy are inappropriate because the pathology is a systemic genetic cartilage disorder, not avascular necrosis.
Q: A child with short stature and epiphyseal irregularities has significant platyspondyly on spine X-rays. Is this consistent with MED? A: No. Significant platyspondyly is not a feature of MED and should prompt consideration of spondyloepiphyseal dysplasia (SED). In MED, the spine is characteristically normal or near-normal. This is one of the most important discriminating features between MED and SED.
Exam Viva Scenarios
Practise clinical reasoning and management decisions out loud
“A 6-year-old boy presents with a waddling gait and bilateral hip pain for 18 months. His father had bilateral hip replacements at age 42. Pelvis X-ray shows bilateral small, fragmented femoral heads with preserved acetabular coverage. Knee X-rays also show small, irregular distal femoral and proximal tibial epiphyses. What is the diagnosis, and how would you manage this child?”
“A 32-year-old woman with known MED (COMP mutation confirmed) presents with end-stage right hip osteoarthritis. She has severe pain limiting her to less than two blocks of walking, night pain, and difficulty with stairs. She works as a schoolteacher. X-rays show complete loss of joint space, subchondral cysts, and osteophytes in the right hip. The left hip has moderate OA with maintained joint space. She is seeking advice about her surgical options. How would you manage her?”
Definition and Genetics
- Skeletal dysplasia with small, irregular epiphyses at multiple joints; spine spared
- Most common gene: COMP (chromosome 19, AD) — approximately 50 percent of cases
- Also COL9A1-3 (AD or AR), MATN3 (AD, milder), SLC26A2 (AR)
- COMP mutations cause ER stress and premature chondrocyte apoptosis
- Allelic with pseudoachondroplasia (same gene, more severe phenotype)
Clinical Presentation
- Waddling gait + joint pain + short stature, onset age 2-10 years
- Hips most commonly affected, then knees, ankles, shoulders
- Spine normal — significant platyspondyly suggests SED, not MED
- Family history often positive (AD) — ask about early arthritis or joint replacements
- Stature 2-3 SD below mean (proportionate, not as severe as achondroplasia)
Diagnosis
- Skeletal survey: small, irregular, delayed epiphyses at multiple joints bilaterally
- Preserved acetabular coverage (distinguishes from Perthes)
- Genetic panel: COMP, COL9A1-3, MATN3 — confirms but not required for management
- Blood tests normal (done to exclude metabolic mimics like hypothyroidism)
- MRI for pre-operative planning, not diagnosis
MED vs Bilateral Perthes
- MED: symmetric, simultaneous, multi-joint, family history, preserved acetabulum
- Perthes: sequential, hip-only, sporadic, acetabular changes develop
- Always check knee X-rays — multi-joint involvement = MED
- Treatment differs: MED is supportive; Perthes may need containment surgery
Management
- Childhood: non-operative — activity modification, physiotherapy, NSAIDs, annual monitoring
- Avoid Perthes-type containment treatment (inappropriate for genetic cartilage disorder)
- Osteotomy only for angular deformity greater than 15 degrees with mechanical axis deviation
- Adult: conservative until end-stage OA, then THA/TKA with durable bearings
- Counsel on young-age arthroplasty: higher revision risk, activity restriction, bilateral planning
Evidence Base and Key Trials
Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene
- First identification of COMP mutations as the cause of MED type 1 (EDM1)
- COMP gene mapped to chromosome 19p13.1
- Demonstrated that COMP mutations cause both MED and pseudoachondroplasia (allelic disorders on a severity spectrum)
- Established the paradigm of cartilage extracellular matrix protein mutations causing skeletal dysplasias
A mutation in the gene encoding the alpha 2 chain of the fibril-associated collagen IX, COL9A2, causes multiple epiphyseal dysplasia (EDM2)
- Identified COL9A2 mutation as the cause of MED type 2 (EDM2)
- Collagen IX is a heterotrimer cross-linking collagen II fibrils in cartilage
- Mutations in COL9 genes produce a generally milder phenotype than COMP mutations
- Established that defects in cartilage collagen fibril architecture, not just matrix proteins, cause MED
Development of the hip in multiple epiphyseal dysplasia. Natural history and susceptibility to premature osteoarthritis
- Reviewed 34 patients with MED, documenting the natural history and radiographic progression
- Hip involvement was universal; knees and ankles were affected in the majority
- Spine was normal or showed only minimal change in all patients
- Early-onset osteoarthritis was the dominant long-term problem, with most patients symptomatic by the third decade
Clinical outcomes of total hip arthroplasty in patients with multiple epiphyseal dysplasia: a single centre study of eighty eight hips at a mean of sixteen year follow-up
- Largest single-centre series of THA in MED patients (88 hips) with mean 16-year follow-up
- THA in MED patients is typically performed at a younger age than for primary OA
- Surgical challenges include altered femoral geometry (short neck, narrow canal) and reduced bone quality
- Durable bearing surfaces recommended given young age and likelihood of future revision
Mutations in the region encoding the von Willebrand factor A domain of matrilin-3 are associated with multiple epiphyseal dysplasia
- Identified MATN3 mutations as the cause of MED type 5 (EDM5), mapped to chromosome 2p24
- MATN3-MED has a milder phenotype with later onset of symptoms and OA
- Matrilin-3 is an extracellular matrix protein involved in cartilage collagen fibril organization
- Expanded the genetic spectrum of MED beyond COMP and COL9