Skeletal-Muscle Differentiated Sarcoma | Embryonal vs Alveolar | Fusion Status Drives Risk
- Most common soft tissue sarcoma in children, arising from cells committed to skeletal-muscle differentiation
- Two main subtypes: embryonal (better prognosis) and alveolar (worse prognosis)
- PAX3-FOXO1 or PAX7-FOXO1 fusion status, not just histology, now drives risk stratification and outcome
- Risk grouping uses pre-treatment stage (TNM site/size/nodes), surgico-pathological clinical group, histology and fusion status
- Treatment is always multimodal: systemic chemotherapy for every patient, plus local control with surgery and/or radiotherapy
- “Fusion-negative alveolar RMS behaves like embryonal RMS - fusion status outperforms morphology
- “Desmin, myogenin (MYF4) and MyoD1 confirm skeletal-muscle differentiation on immunohistochemistry
- “Parameningeal, extremity and unfavourable-site tumours and any nodal/metastatic disease worsen risk
- “Adult and pleomorphic rhabdomyosarcoma carry a markedly worse prognosis than paediatric disease
Rhabdomyosarcoma is the malignant counterpart of skeletal muscle, even when it arises where there is no striated muscle (bladder, biliary tree, orbit). It is the most common soft tissue sarcoma of childhood.
PAX3-FOXO1 and PAX7-FOXO1 fusions define the high-risk group. Fusion-negative alveolar tumours behave like embryonal disease. Always request fusion testing - it changes risk allocation.
Every patient receives systemic chemotherapy (vincristine, actinomycin D, cyclophosphamide backbone) plus local control with surgery and/or radiotherapy. Surgery alone is never adequate.
Primary site, tumour size, nodal status and resectability all feed risk grouping. Parameningeal and extremity sites and any metastatic disease carry the worst outlook.
- Answer
- Yes - rhabdomyosarcoma
- Answer
- Cells committed to skeletal-muscle differentiation
- Answer
- Embryonal (better) and alveolar (worse)
- Answer
- PAX3-FOXO1 or PAX7-FOXO1 fusion
- Answer
- Desmin, myogenin (MYF4), MyoD1
- Answer
- Systemic chemotherapy (VAC backbone) for every patient
- Answer
- Surgery and/or radiotherapy, risk-adapted
- Answer
- Metastatic, fusion-positive, alveolar, and adult disease
MUSCLESRhabdomyosarcoma Key Features
Hook:MUSCLES - because rhabdomyosarcoma is the cancer of skeletal muscle differentiation.
EAEmbryonal vs Alveolar Subtypes
Hook:E before A - Embryonal is Easier (better prognosis) than Alveolar.
FAILSAdverse Prognostic Factors
Hook:When these factors stack up, treatment more often FAILS.
Overview and Epidemiology
Rhabdomyosarcoma (RMS) is a malignant soft tissue tumour whose cells are committed to skeletal-muscle differentiation. It is the most common soft tissue sarcoma of childhood and one of the more frequent solid tumours in children overall. Despite its skeletal-muscle phenotype, it can arise anywhere - including sites that contain no striated muscle, such as the bladder, biliary tree and orbit.
Rhabdomyosarcoma does not require pre-existing skeletal muscle to form. It arises from primitive mesenchymal cells that activate the skeletal-muscle differentiation programme (MyoD1, myogenin). This is why a bladder or orbital tumour can still be a rhabdomyosarcoma.
- Age: Most cases under 10 years; a smaller second peak in adolescence
- Bimodal biology: Embryonal dominates in young children, alveolar in older children and teens
- Adults: Rare and carry a markedly worse prognosis
- Slight male predominance overall
- Head and neck (including orbit and parameningeal): most common region
- Genitourinary (bladder, prostate, paratesticular, vagina)
- Extremity: more often alveolar, higher nodal risk
- Trunk, retroperitoneum and other sites
Favourable vs Unfavourable Primary Sites
- Examples
- Orbit, non-parameningeal head and neck, genitourinary non-bladder/prostate (e.g. paratesticular, vagina), biliary tract
- Prognostic Significance
- Better outcome; allow treatment de-escalation in low-risk groups
- Examples
- Parameningeal, bladder/prostate, extremity, trunk, retroperitoneum
- Prognostic Significance
- Higher relapse and metastatic risk; require intensified therapy
Rhabdomyosarcoma in adults is uncommon and has a substantially poorer prognosis than the paediatric disease, partly because of more frequent pleomorphic histology, unfavourable sites and later presentation. Extremity rhabdomyosarcoma - the form most relevant to orthopaedic practice - is frequently alveolar and carries a higher rate of nodal involvement.
Pathophysiology and Molecular Biology
Cellular Origin and the Myogenic Programme
Rhabdomyosarcoma cells express the master regulators of skeletal-muscle differentiation - MyoD1 and myogenin (MYF4) - together with structural muscle proteins such as desmin. This myogenic signature is the basis for diagnosis on immunohistochemistry.
Defined by gene fusions involving FOXO1:
- PAX3-FOXO1 (t(2;13)) - most common, worst prognosis
- PAX7-FOXO1 (t(1;13)) - less common, somewhat better behaviour
- Drives an aggressive transcriptional programme
- Most morphologically alveolar tumours are fusion-positive
No FOXO1 fusion; driven by other mutations:
- RAS pathway mutations are common
- Includes most embryonal tumours
- Generally better prognosis than fusion-positive disease
- Fusion-negative alveolar tumours behave like embryonal RMS
Modern risk stratification is increasingly based on FOXO1 fusion status rather than alveolar versus embryonal morphology alone. Large cooperative-group data show that alveolar tumours without a fusion behave like embryonal disease, while fusion-positive tumours behave aggressively regardless of subtle morphology. Always request molecular fusion testing.
Other Recurrent Molecular Drivers
- Typical Context
- Fusion-negative (mostly embryonal) tumours
- Clinical Relevance
- Found in over half of fusion-negative cases; potential therapeutic target
- Typical Context
- Both fusion-positive and fusion-negative tumours
- Clinical Relevance
- Associated with worse outcome; flags possible Li-Fraumeni predisposition
- Typical Context
- Spindle cell/sclerosing RMS, often older patients
- Clinical Relevance
- Highly aggressive subset despite spindle morphology
- Typical Context
- Congenital/infantile spindle cell RMS
- Clinical Relevance
- Generally favourable behaviour in infants
A minority of rhabdomyosarcomas arise in cancer predisposition syndromes - Li-Fraumeni (TP53), neurofibromatosis type 1, DICER1, Costello and Beckwith-Wiedemann syndromes. Young age, embryonal/anaplastic histology and a relevant family history should prompt consideration of genetic referral.
Classification and Histology
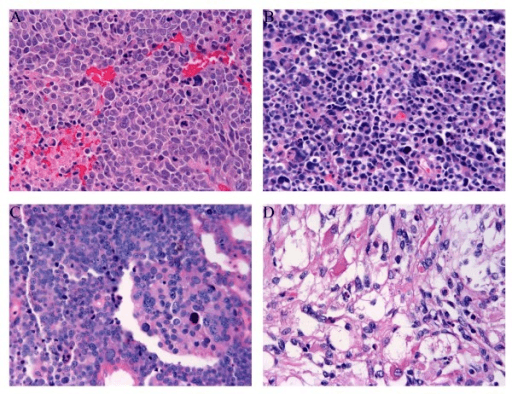
WHO Histological Subtypes
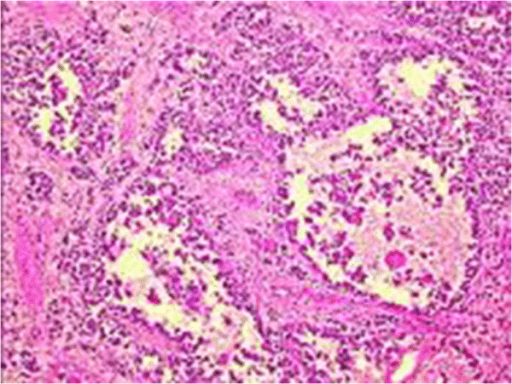
Embryonal Rhabdomyosarcoma (ERMS)
The most common subtype, typically in younger children and at head/neck and genitourinary sites.
- Primitive small round to spindle cells in a myxoid stroma
- Variable rhabdomyoblasts with eosinophilic cytoplasm (tadpole/strap cells)
- Alternating cellular and loose hypocellular areas
- Botryoid variant: grape-like polypoid growth under mucosa (e.g. bladder, vagina), with a cambium layer
- Usually FOXO1 fusion-negative
- Frequent RAS pathway mutations
- Generally favourable prognosis
- Botryoid and spindle-cell variants are favourable
Embryonal histology, especially at a favourable site, allows treatment de-escalation in the lowest-risk groups.
Immunohistochemistry
- Significance
- Muscle intermediate filament
- Diagnostic Value
- Sensitive but not specific for skeletal muscle
- Significance
- Skeletal-muscle transcription factor
- Diagnostic Value
- Specific; diffuse strong staining favours alveolar subtype
- Significance
- Master myogenic regulator
- Diagnostic Value
- Specific for skeletal-muscle differentiation; key in spindle/sclerosing RMS
- Significance
- Detects PAX-FOXO1 fusion
- Diagnostic Value
- Defines fusion-positive (high-risk) disease
Diffuse, strong nuclear myogenin staining is typical of alveolar (often fusion-positive) tumours, whereas embryonal tumours tend to show more patchy myogenin. Combine immunohistochemistry with molecular fusion testing for definitive classification.
On low power, RMS is a small round blue cell tumour (SRBCT) - densely packed primitive cells with scant cytoplasm - and examiners use it to test the paediatric SRBCT differential, because the management of each member is completely different. You distinguish them by immunohistochemistry and molecular genetics, not morphology:
- Rhabdomyosarcoma: desmin, myogenin and MyoD1 (nuclear) positive; PAX3/PAX7-FOXO1 fusion in alveolar - the only one with skeletal-muscle markers.
- Ewing sarcoma / PNET: diffuse membranous CD99, NKX2.2; EWSR1-FLI1 (t(11;22)) fusion - muscle markers negative.
- Neuroblastoma: young child, raised urinary catecholamines (VMA/HVA), PHOX2B, NSE, synaptophysin; Homer-Wright rosettes; MYCN amplification.
- Lymphoblastic lymphoma / leukaemia: LCA (CD45), TdT and lineage markers positive.
- Desmoplastic small round cell tumour (DSRCT): abdominal/peritoneal, polyphenotypic (desmin dot-like, keratin, WT1) with EWSR1-WT1 fusion - do not mistake the desmin positivity for RMS.
- Wilms tumour (blastemal), and in bone the mesenchymal chondrosarcoma / small cell osteosarcoma also enter the list.
Exam point: a paediatric SRBCT is not a diagnosis - RMS is confirmed by desmin/myogenin/MyoD1 and (for alveolar) FOXO1 fusion, separating it from the CD99-positive Ewing, PHOX2B-positive neuroblastoma, LCA/TdT-positive lymphoma, and EWSR1-WT1 DSRCT.
Clinical Presentation
History
- Painless or painful enlarging mass at the primary site
- Site-specific symptoms: proptosis (orbit), nasal obstruction or cranial nerve palsy (parameningeal), haematuria/urinary obstruction (bladder), painless scrotal mass (paratesticular)
- Rapid growth is common
- Constitutional symptoms suggest metastatic disease
- Cranial nerve palsy or meningeal symptoms (parameningeal extension)
- A deep, firm, enlarging extremity mass in a child or adolescent
- Regional lymphadenopathy (especially extremity and paratesticular)
- Bone pain or pancytopenia (marrow involvement)
A deep, enlarging or unexplained soft tissue mass in a child or young adult must be imaged and biopsied at a sarcoma centre rather than excised locally. As with all sarcomas, an unplanned excision compromises subsequent margins and contaminates tissue planes.
Physical Examination
Examination Approach
- Size and site of the mass
- Skin changes, overlying vascularity, proptosis or facial asymmetry
- Functional impairment of the adjacent joint or organ
- Consistency, fixity and relation to deep structures
- Tenderness and neurovascular relationship
- Regional lymph node assessment (important for extremity and paratesticular tumours)
- Cranial nerve examination for head/neck tumours
- Distal neurovascular status for extremity tumours
- General examination for hepatosplenomegaly, bone tenderness and signs of marrow failure
Investigations and Imaging
Imaging and Staging Workup
Local Imaging
MRI is the standard for local staging of the primary tumour, defining size, compartment, and relationship to neurovascular structures and bone.
- MRI: T1, fluid-sensitive sequences and post-contrast imaging of the whole compartment
- CT: useful for bony involvement and for head/neck base-of-skull assessment
- Ultrasound: initial assessment of superficial and scrotal masses
Accurate local imaging underpins both the TNM stage and surgical/radiotherapy planning.
Biopsy Principles
Apply standard sarcoma biopsy principles:
- Refer to a sarcoma multidisciplinary centre before biopsy
- Core needle biopsy preferred; plan the tract so it can be excised at definitive surgery
- Use a longitudinal approach for extremity tumours; avoid transverse incisions and contamination of separate compartments
- Obtain adequate tissue for histology, immunohistochemistry and molecular fusion testing
- Never perform an unplanned excisional biopsy of an undiagnosed deep mass
Staging and Risk Grouping
Rhabdomyosarcoma uses a combination of systems: a pre-treatment TNM stage (based on site, tumour size and invasiveness, nodal status and metastases), a surgico-pathological clinical group (extent of residual disease after initial surgery), histology and fusion status. These combine into low-, intermediate- and high-risk groups that determine therapy.
- Definition
- Localised tumour, completely resected, negative margins
- Implication
- Lowest local-treatment burden
- Definition
- Gross total resection with microscopic residual and/or resected positive nodes
- Implication
- Local radiotherapy usually added
- Definition
- Gross residual disease after biopsy or incomplete resection
- Implication
- Most common group; needs radiotherapy and chemotherapy
- Definition
- Distant metastatic disease at diagnosis
- Implication
- High-risk; intensified systemic therapy
- Favourable
- Embryonal or fusion-negative
- Unfavourable
- Fusion-positive (PAX3-FOXO1 worst)
- Favourable
- Orbit, non-parameningeal head/neck, favourable GU
- Unfavourable
- Parameningeal, extremity, bladder/prostate, trunk
- Favourable
- Smaller, non-invasive (T1)
- Unfavourable
- Over 5cm, invasive (T2)
- Favourable
- Node-negative, non-metastatic
- Unfavourable
- Node-positive or metastatic
- Favourable
- Group I-II
- Unfavourable
- Group III-IV
Management
Core Principles
Every patient with rhabdomyosarcoma receives systemic chemotherapy, combined with local control by surgery and/or radiotherapy. The intensity and duration are tailored to the risk group. Management is delivered by a paediatric/sarcoma multidisciplinary team.
Treatment Fundamentals:
- Chemotherapy: vincristine, actinomycin D (dactinomycin) and cyclophosphamide (VAC) form the backbone; ifosfamide-containing and other regimens are used in different cooperative groups, with intensification for higher risk
- Surgery: complete resection where it can be achieved without unacceptable morbidity; otherwise biopsy followed by chemotherapy and delayed local control
- Radiotherapy: for microscopic or gross residual disease, nodal involvement and most alveolar tumours
These principles apply across all sites, including extremity disease relevant to orthopaedic surgeons.
A defining feature of rhabdomyosarcoma versus adult soft-tissue sarcoma is how central radiotherapy is to local control. Because chemotherapy is universal and complete excision is often unachievable without mutilation, almost every RMS patient receives radiotherapy - the principal exception is the patient with completely resected, group I embryonal disease at a favourable site, who may avoid it. Microscopic residual (group II), gross residual (group III), nodal disease, and essentially all alveolar/fusion-positive tumours get radiotherapy.
The second examinable point is how it is delivered in a growing child, because the late effects (growth arrest, asymmetry, organ damage, second malignancy) are lifelong:
- Proton beam therapy is increasingly used to reduce dose to adjacent growing bone, brain (parameningeal/orbital tumours) and viscera.
- Brachytherapy (e.g. for selected genitourinary/vaginal and small extremity tumours) delivers a high local dose over a small volume, sparing surrounding growth plates and organs.
- Function- and growth-sparing planning, not dose escalation, is the priority.
Exam point: in RMS, radiotherapy is the rule, not the exception (only completely-resected group I embryonal may skip it), and in children modality choice (proton beam, brachytherapy, conformal planning) is driven by minimising lifelong growth and late-toxicity damage.
Surgical Considerations
Surgical Role in Rhabdomyosarcoma
Unlike adult extremity soft tissue sarcomas where surgery is often the primary treatment, in rhabdomyosarcoma chemotherapy is universal and surgery is integrated into a multimodal plan.
Decision Sequence
- Considered when complete excision with negative margins is achievable without major functional loss or mutilation
- Appropriate for small, favourable, resectable tumours (e.g. paratesticular, some extremity lesions)
- Avoids or reduces radiotherapy in selected patients
- For larger or unfavourable-site tumours, perform diagnostic biopsy only
- Give neoadjuvant chemotherapy to shrink the tumour
- Reassess for delayed primary resection or definitive radiotherapy
- Delayed primary excision can convert a group III tumour to a resected state
- Radiotherapy is used when surgery would be mutilating or margins remain positive
- Function preservation is prioritised, especially in children
Complications
Disease and Treatment-Related Complications
- Context
- Incomplete margins, gross residual disease, high-risk biology
- Management / Mitigation
- Re-resection if feasible, radiotherapy, systemic therapy
- Context
- Lung, bone, bone marrow; more common in alveolar/fusion-positive disease
- Management / Mitigation
- Intensified systemic therapy, clinical trials, selective metastasis-directed treatment
- Context
- Cyclophosphamide and ifosfamide (gonadotoxicity, haemorrhagic cystitis), anthracycline cardiotoxicity, myelosuppression
- Management / Mitigation
- Dose tailoring, mesna, fertility counselling, cardiac monitoring
- Context
- Growth disturbance, fibrosis, organ-specific damage, second malignancy
- Management / Mitigation
- Modern conformal techniques, minimise dose/field, long-term surveillance
- Context
- Functional loss, nerve injury, wound problems
- Management / Mitigation
- Limb-sparing planning, reconstruction, rehabilitation
Because many children are cured, the long-term toxicity of treatment (infertility, cardiac dysfunction, growth problems and second cancers) is a central concern. This drives the trend towards risk-adapted de-escalation in low-risk patients and structured long-term survivor follow-up.
Prognosis and Outcomes
Prognostic Factors
- Favourable
- Fusion-negative
- Unfavourable
- PAX3-FOXO1 (worst), PAX7-FOXO1
- Favourable
- Embryonal (incl. botryoid/spindle variants)
- Unfavourable
- Alveolar; anaplasia in embryonal tumours
- Favourable
- Orbit, favourable head/neck, favourable GU
- Unfavourable
- Parameningeal, extremity, bladder/prostate, trunk
- Favourable
- Localised, node-negative
- Unfavourable
- Node-positive or metastatic
- Favourable
- Group I-II
- Unfavourable
- Group III-IV
- Favourable
- 1-9 years
- Unfavourable
- Under 1 or over 10 years; adults much worse
Among morphologically alveolar and embryonal tumours treated with the same intermediate-risk therapy, fusion-positive disease (particularly PAX3-FOXO1) has significantly worse event-free and overall survival, while fusion-negative alveolar disease behaves like embryonal RMS. This is why fusion status is now embedded in risk stratification.
Survival Overview
- Outlook
- High cure rates with risk-adapted therapy
- Outlook
- Intermediate survival; benefits from full multimodal therapy
- Outlook
- Poor survival despite intensified treatment
- Outlook
- Markedly worse than paediatric disease
Clinical Relevance and Controversies
The shift from histology-based to fusion-based risk stratification, supported by international genomic data, is refining who needs intensification versus de-escalation. Incorporating additional drivers (e.g. MYOD1, TP53) into routine risk grouping is an active area of trial design.
Reducing cyclophosphamide and shortening therapy in low-risk patients lowers late toxicity, but the balance between minimising harm and maintaining cure rates must be confirmed in each subgroup before being adopted as standard.
Adults are frequently treated on paediatric-style protocols, and those who receive intensive multimodal therapy fare better than historical adult cohorts, yet outcomes remain inferior to children. Optimal regimens for adults are still debated.
Outcomes after relapse, and for metastatic disease at presentation, remain poor. New agents (targeted therapy against RAS pathway, immunotherapy and antibody-based approaches) are under investigation, and trial enrolment is encouraged.
Guidelines, Registries & Global Practice
Global Epidemiology
- Most common soft tissue sarcoma of childhood worldwide
- Majority of cases occur under 10 years, with a second adolescent peak
- Embryonal subtype predominates in young children; alveolar in older children/teens
- Adults are rarely affected and have markedly worse outcomes
- Common metastatic sites are lung, regional nodes, bone and bone marrow
- Outcome ranges from high cure rates in low-risk disease to poor survival when metastatic
- Fusion-positive biology is a key global driver of poor outcome
Cooperative-Group Frameworks
- Risk Stratification
- Stage, clinical group, histology and increasingly fusion status
- Chemotherapy Backbone
- Vincristine, actinomycin D, cyclophosphamide (VAC) and intensified variants
- Emphasis
- De-escalation in low-risk; intensification and trials in high-risk
- Risk Stratification
- Risk-adapted protocols using site, size, nodes, age and fusion status
- Chemotherapy Backbone
- Ifosfamide-, vincristine- and actinomycin-based regimens with maintenance therapy
- Emphasis
- Function-preserving local control and maintenance chemotherapy
- Risk Stratification
- Mandatory specialist multidisciplinary review
- Chemotherapy Backbone
- Protocol-based multi-agent chemotherapy
- Emphasis
- Centralised diagnosis, molecular confirmation and treatment
Regardless of region, all frameworks agree on three points: any suspicious deep or enlarging soft tissue mass should be referred to a specialist sarcoma centre before biopsy, diagnosis should be confirmed histologically and molecularly (including FOXO1 fusion status), and treatment should be multimodal, risk-adapted and delivered by a multidisciplinary team.
High- vs Limited-Resource Practice Variation
- Routine FOXO1 fusion testing and broader molecular profiling
- Centralised paediatric sarcoma multidisciplinary teams
- Modern conformal radiotherapy and limb-sparing surgery
- Structured survivorship and late-effects surveillance
- Diagnosis may rely on morphology and immunohistochemistry where molecular testing is unavailable
- Treatment refusal and abandonment can materially reduce survival
- Later presentation with larger, unfavourable-site tumours
- Regional referral networks and protocol adaptation help bridge gaps
Applicable in any health system:
- Document the investigation pathway for any persistent deep soft tissue mass
- Confirm pre-biopsy staging and that biopsy was planned with the definitive surgeon
- Record that histology, immunohistochemistry and FOXO1 fusion testing were requested
- Evidence of sarcoma multidisciplinary discussion before definitive treatment
- Informed consent covering multimodal therapy, fertility and long-term toxicity
- A written long-term survivorship and surveillance plan
MCQ Practice Points
Q: What is the most common soft tissue sarcoma of childhood? A: Rhabdomyosarcoma. It arises from cells committed to skeletal-muscle differentiation and most often affects children under 10 years, with a smaller second peak in adolescence.
Q: Which gene fusions define high-risk alveolar rhabdomyosarcoma? A: PAX3-FOXO1 (t(2;13)) and PAX7-FOXO1 (t(1;13)). PAX3-FOXO1 carries the worst prognosis. Fusion-negative alveolar tumours behave like embryonal disease.
Q: Which immunohistochemical markers confirm rhabdomyosarcoma? A: Desmin, myogenin (MYF4) and MyoD1. Myogenin and MyoD1 are nuclear skeletal-muscle transcription factors; diffuse strong myogenin staining favours the alveolar subtype.
Q: What is the universal component of rhabdomyosarcoma treatment? A: Systemic chemotherapy - typically a vincristine, actinomycin D and cyclophosphamide (VAC) backbone - given to every patient, combined with local control by surgery and/or radiotherapy and tailored to risk group.
Q: Name key adverse prognostic factors in rhabdomyosarcoma. A: Fusion-positive (especially PAX3-FOXO1) disease, alveolar histology or anaplasia, unfavourable primary site, large/invasive tumour, nodal or metastatic spread, higher clinical group, and adult age.
Exam Viva Scenarios
Practise clinical reasoning and management decisions out loud
“A 14-year-old presents with a firm, deep 6cm mass in the anterior thigh that has grown over two months. There is a palpable inguinal node. How do you investigate and what is your differential?”
“A child's biopsy is reported as alveolar rhabdomyosarcoma. The treating oncologist asks whether molecular testing will change anything. How do you respond?”
“A young child has a completely resected paratesticular embryonal rhabdomyosarcoma that is fusion-negative and node-negative. The parents are worried about chemotherapy toxicity. How do you counsel them and plan treatment?”
Key Epidemiology
- Most common soft tissue sarcoma of childhood
- Majority under 10 years; second adolescent peak
- Common sites: head and neck (incl. orbit, parameningeal), genitourinary, extremity, trunk
- Adult and pleomorphic disease is rare and has a much worse prognosis
Histology and Molecular Biology
- Embryonal (commonest, better prognosis, usually fusion-negative)
- Alveolar (older patients, extremity/trunk, often PAX-FOXO1 fusion-positive, worse prognosis)
- Pleomorphic (adults) and spindle/sclerosing (MYOD1-mutant aggressive) variants
- PAX3-FOXO1 and PAX7-FOXO1 fusions define high-risk biology; fusion status now drives risk
Diagnosis
- Refer to sarcoma centre before biopsy; planned excisable tract
- Immunohistochemistry: desmin, myogenin (MYF4), MyoD1
- FOXO1 fusion testing (FISH/RT-PCR) on every case
- MRI for local staging; CT chest, whole-body imaging and bone marrow for distant staging
Risk Grouping
- Pre-treatment TNM stage (site, size, invasiveness, nodes, metastases)
- Surgico-pathological clinical group I-IV (residual disease)
- Histology and fusion status
- Combine into low-, intermediate- and high-risk groups
Treatment (Always Multimodal)
- Systemic chemotherapy for every patient (VAC backbone and variants)
- Local control with surgery and/or radiotherapy
- Upfront resection only if complete and non-mutilating; otherwise biopsy then chemo then delayed local control
- Low-risk de-escalation: shorter therapy, reduced/omitted cyclophosphamide, lower radiotherapy dose
Adverse Prognostic Factors (FAILS)
- Fusion-positive (PAX3-FOXO1 worst)
- Alveolar histology and anaplasia
- Invasive or large tumour (over 5cm, T2)
- Lymph node or metastatic spread
- Unfavourable Site and age (under 1 or over 10 years; adults much worse)
Complications and Survivorship
- Local recurrence and distant metastases (lung, bone, marrow)
- Chemotherapy toxicity: gonadotoxicity, haemorrhagic cystitis, cardiotoxicity
- Radiotherapy late effects: growth disturbance, fibrosis, second malignancy
- Long-term survivor surveillance is essential, especially in cured children
Evidence Base and Key Studies
PAX-FOXO1 Fusion Status Drives Outcome in Intermediate-Risk RMS
- Children's Oncology Group D9803 cohort; 434 cases with full clinical, molecular and pathology data
- Event-free survival was worse for alveolar PAX3-FOXO1 (54%) and PAX7-FOXO1 (65%) than embryonal RMS (77%)
- Fusion-negative alveolar RMS had outcomes similar to embryonal RMS (90% vs 77% EFS, not significantly different)
- PAX3-FOXO1 tumours had the poorest overall survival (64%) compared with PAX7-FOXO1, fusion-negative alveolar and embryonal disease
Genomic Classification and Clinical Outcome in Rhabdomyosarcoma
- International consortium; custom-capture sequencing of 641 rhabdomyosarcoma tumours from COG and UK trials
- In fusion-negative cases, RAS pathway mutations were present in over half, while about 21% had no identified driver
- TP53 mutations were associated with worse outcome in both fusion-negative and fusion-positive disease
- MYOD1 mutation was linked to older age, head and neck primaries and a dismal survival, and is being incorporated into risk stratification
Anaplasia as a Prognostic Factor in Childhood RMS
- Prospective assessment of anaplasia in 546 children on IRSG/COG trials (1995-1998)
- Anaplasia (focal or diffuse) was present in 13% of samples - more common than previously reported
- In embryonal RMS, anaplasia reduced 5-year failure-free survival (63% vs 77%) and overall survival (68% vs 82%)
- The adverse effect was most pronounced in intermediate-risk embryonal tumours; anaplasia did not affect alveolar tumour outcome