Periarticular Hydroxyapatite Masses | FGF23/GALNT3 Pathway | Hip, Shoulder, Elbow
- Periarticular lobulated masses of calcium hydroxyapatite, not a true neoplasm
- Hip, shoulder, elbow and wrist are the classic sites; mass often grows over years
- Hyperphosphataemic type = GALNT3, FGF23 or KLOTHO loss-of-function - phosphate retention
- Secondary type is far more common than familial, and is driven by chronic renal failure
- X-ray: lobulated, 'cloud-like' or 'chicken-wire' periarticular calcification is characteristic
- Biopsy is usually diagnostic; differentiate from parosteal osteosarcoma, myositis ossificans, tophaceous gout
- “Serum calcium is normal, phosphate is high (in familial hyperphosphataemic type) - 1,25-(OH)2 vitamin D is suppressed
- “Chalky white 'toothpaste' discharge through skin is pathognomonic in ulcerated cases
- “Myositis ossificans forms mature lamellar bone attached to periosteum - tumoral calcinosis does not
- “Recurrence after excision is the rule, not the exception, in familial disease
- “Acetazolamide and phosphate binders (sevelamer, lanthanum) are first-line metabolic therapy
Periarticular calcium hydroxyapatite deposition. In familial (primary hyperphosphataemic) disease, loss-of-function mutations in GALNT3, FGF23 or KLOTHO impair FGF23 signalling, causing renal phosphate retention and hyperphosphataemia. The deposited mineral is calcium phosphate (hydroxyapatite), not calcium oxalate or calcium pyrophosphate.
Hip greater than shoulder greater than elbow greater than wrist. The masses sit adjacent to but not within the joint, often along the trochanteric bursa, subdeltoid bursa, or olecranon. Lesions are lobulated, can exceed 20 cm, and may ulcerate through skin discharging chalky material.
Hyperphosphataemic familial (FGF23 axis) vs normophosphataemic primary vs secondary uraemic vs dystrophic. Always check serum calcium, phosphate, PTH, 25-OH vitamin D, 1,25-(OH)2 vitamin D, creatinine, and urinary phosphate. Familial disease typically presents in the 1st-2nd decade.
Metabolic first, surgery for symptoms. Phosphate binders (sevelamer, lanthanum), low-phosphate diet, and acetazolamide reduce phosphate load. Surgical excision is reserved for symptomatic, ulcerating, mechanically limiting or diagnostically uncertain masses. Recurrence is the rule in familial disease, often approaching 100 percent after simple debulking.
- Phosphate
- Elevated (often greater than 2 mmol/L)
- Clinical Clue
- Child/young adult, sibling cases, no renal disease
- First-Line Treatment
- Phosphate binder + acetazolamide + low-P diet
- Surgical Prognosis
- High recurrence (often greater than 50%)
- Phosphate
- Normal
- Clinical Clue
- Solitary mass, no metabolic abnormality
- First-Line Treatment
- Excision if symptomatic
- Surgical Prognosis
- Low recurrence if completely excised
- Phosphate
- Often elevated, with high PTH and Ca-P product
- Clinical Clue
- Adult on long-term dialysis, multiple joints
- First-Line Treatment
- Manage CKD-MBD, parathyroidectomy if needed
- Surgical Prognosis
- Recurrence common unless metabolic state corrected
- Phosphate
- Variable
- Clinical Clue
- Local tissue damage or systemic metabolic driver
- First-Line Treatment
- Treat underlying cause, excision of mass
- Surgical Prognosis
- Depends on cause control
PHOSPHATEHyperphosphataemic Familial Type
Hook:PHOSPHATE captures the entire metabolic story of familial tumoral calcinosis.
RECURRINGSurgical and Prognostic Pearls
Hook:Tumoral calcinosis is RECURRING - prevention is the only cure for the familial form.
Overview and Epidemiology
Tumoral calcinosis is a frequently missed cause of a periarticular 'tumour' in young patients. It is not a neoplasm - it is a metabolic deposition of calcium hydroxyapatite, almost always driven by either an inherited FGF23-axis defect (rare, familial, paediatric) or chronic renal failure with disordered mineral metabolism (common, adult, dialysis-dependent). A correct diagnosis changes management completely: surgery cures neither the familial form (it always recurs) nor the uraemic form (unless the metabolic state is corrected).
- Rare disease: overall incidence estimated at roughly 1 in 100,000 to 1 in 1,000,000
- Familial (primary hyperphosphataemic): autosomal recessive, presents in 1st-2nd decade, often in children of consanguineous parents; described in clusters from the Middle East, North Africa and Turkey
- Secondary (uraemic): the clinically dominant form, rising in prevalence with the dialysis population
- Endemic reports: high-frequency clusters described in parts of sub-Saharan Africa and Papua New Guinea, often associated with low calcium, high phosphate dietary patterns
- Sex: familial form shows no strong sex predilection; secondary form tracks with CKD demographics
- Diagnostic confusion with bone or soft-tissue tumour - biopsy or excision is the typical referral pathway
- Mechanical symptoms - large masses restrict joint motion, irritate bursa, compress nerves
- Skin breakdown - ulceration with chalky 'toothpaste' discharge (pathognomonic when present)
- Functional limitation - sitting, walking, dressing all impaired
- Recurrence after incomplete excision - the single most common reason for repeated referrals
Pathophysiology
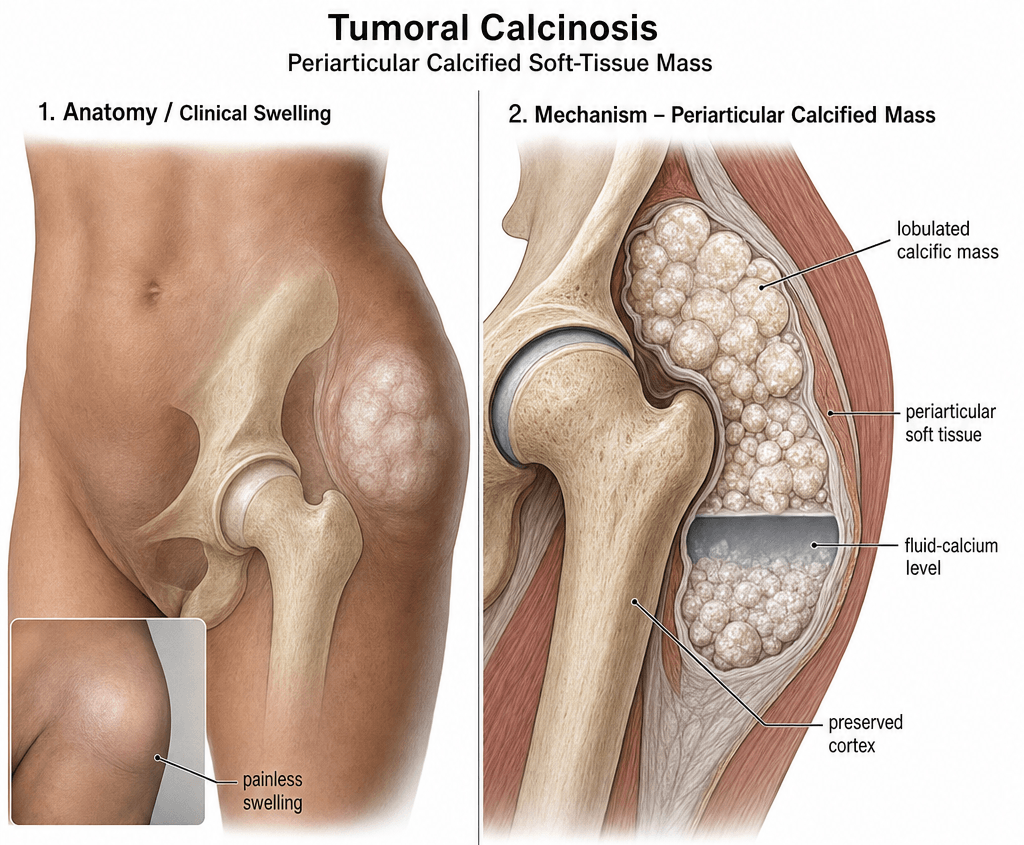
The mineral is calcium hydroxyapatite (calcium phosphate), deposited in lobulated sheets within periarticular soft tissue, often with a foreign-body giant cell reaction. There is no cellular atypia. The mass is not encapsulated and tends to insinuate around tendons, bursae and neurovascular structures, which is what makes complete excision surgically challenging and recurrence common.
- Driver
- GALNT3, FGF23 or KLOTHO loss-of-function - renal phosphate retention
- Lab Pattern
- High PO4, normal Ca, suppressed 1,25-(OH)2D, normal/low PTH
- Histology
- Lobulated hydroxyapatite with giant cells
- Recurrence Risk
- Very high (often greater than 50%)
- Driver
- Unknown, possibly post-traumatic, local tissue injury
- Lab Pattern
- Normal serum calcium and phosphate
- Histology
- Lobulated hydroxyapatite, often solitary
- Recurrence Risk
- Low if completely excised
- Driver
- Hyperphosphataemia, secondary hyperparathyroidism, high Ca-P product, vitamin D analogues
- Lab Pattern
- High PO4, high PTH, variable Ca, low/normal vitamin D
- Histology
- Multilobulated periarticular calcium deposits
- Recurrence Risk
- Common if metabolic state uncontrolled
- Driver
- Local tissue damage (trauma, surgery) or systemic drivers (hyperparathyroidism, vitamin D toxicity, milk-alkali)
- Lab Pattern
- Depends on cause
- Histology
- Calcification within damaged tissue
- Recurrence Risk
- Depends on cause control
FGF23 is secreted by osteocytes and osteoblasts in response to phosphate load and active vitamin D. Active FGF23 requires O-linked glycosylation by GALNT3 to be secreted in an intact form. KLOTHO is the co-receptor in the kidney.
In the familial hyperphosphataemic type: biallelic loss-of-function of any of these three genes (GALNT3 most common) prevents FGF23 signalling, abolishing renal phosphate wasting and 1-alpha-hydroxylase suppression.
Net result: phosphate retention, hyperphosphataemia, suppressed 1,25-(OH)2 vitamin D, normal calcium, normal or low PTH, and progressive periarticular hydroxyapatite deposition.
CKD-Mineral and Bone Disorder (CKD-MBD) is the umbrella term. As GFR falls below roughly 30 mL/min/1.73 m^2, phosphate excretion fails, FGF23 rises, active vitamin D falls, and PTH rises (secondary hyperparathyroidism).
Drivers of soft-tissue calcification in CKD include: hyperphosphataemia, raised calcium-phosphate product, calciphylaxis-spectrum small-vessel disease, vitamin D analogue dosing, and use of calcium-based phosphate binders.
Tumoral calcinosis lesions in dialysis patients can reach enormous size at the hip, shoulder, elbow, hand or wrist, and frequently ulcerate.
The deposits form at sites of microtrauma, repeated shear or bursal friction - explaining the predilection for the greater trochanteric bursa, subdeltoid bursa, olecranon bursa and ischial region. The combination of local tissue injury, alkaline local pH, and high extracellular phosphate favours hydroxyapatite (rather than calcium oxalate) precipitation. The mineral is rarely within the joint itself.
An examinable extension that this topic must name: hyperphosphataemic familial tumoral calcinosis (HFTC) and hyperostosis-hyperphosphataemia syndrome (HHS) are two phenotypes of ONE disease. HHS is caused by the same biallelic GALNT3 (or FGF23) loss-of-function mutations and shows the identical biochemistry - high phosphate, normal calcium, suppressed 1,25-(OH)2 vitamin D and increased tubular phosphate reabsorption.
Instead of (or as well as) periarticular calcific masses, HHS presents with painful diaphyseal hyperostosis - cortical thickening and periosteal new bone, classically of the long bones (the tibia and ulna are typical), with recurrent episodes of bone pain, local swelling and raised inflammatory markers.
- Why it matters: this presentation is a recognised mimic of chronic recurrent multifocal osteomyelitis (CRMO), osteomyelitis or a bone tumour. Recognising the same hyperphosphataemic biochemistry and the family history prevents misdiagnosis and unnecessary biopsy/antibiotics.
- HFTC and HHS can coexist in the same family or even the same patient, so a child with unexplained diaphyseal hyperostosis and hyperphosphataemia should be worked up for the FGF23-axis defect, and the metabolic phosphate-lowering treatment is the same.
Classification and Types
Pathogenesis-Based Classification (Smack 1996)
- Phosphate
- Elevated, often greater than 2 mmol/L
- Typical Patient
- Child or young adult, autosomal recessive, no renal disease
- Distribution
- Hip, shoulder, elbow; can be multifocal
- Phosphate
- Normal
- Typical Patient
- Any age, often solitary, no systemic disease
- Distribution
- Hip, elbow, hand
- Phosphate
- Elevated, with high PTH and Ca-P product
- Typical Patient
- Adult on long-term dialysis or advanced CKD
- Distribution
- Hip, shoulder, hand, wrist; often multiple sites
- Phosphate
- Variable
- Typical Patient
- Local trauma, hyperparathyroidism, vitamin D toxicity, milk-alkali, connective tissue disease
- Distribution
- Often single, at site of injury or systemic driver
Pathogenesis matters because it dictates whether the disease is curable, controllable or simply manageable.
Clinical Assessment
- Mass duration: years of slow, often painless growth is the rule
- Pain: usually late, when mass irritates bursa, compresses nerve or ulcerates skin
- Discharge: chalky white 'toothpaste' through skin is pathognomonic
- Renal disease: dialysis vintage, transplant status, medications (calcium-based binders, vitamin D analogues)
- Family history: sibling or parental cases (autosomal recessive)
- Diet and drugs: high phosphate intake, vitamin D supplementation, calcium supplements
- Mass: firm, lobulated, 'stony-hard', non-tender, often warm but not inflamed
- Mobility: usually fixed to deep soft tissues, not to skin (until late)
- Skin: tethering, erythema, ulceration, sinus tracts
- Joint: range often preserved unless mass mechanically blocks it
- Neurovascular: document function pre-op (sciatic at hip, axillary at shoulder, ulnar at elbow)
- Lymph nodes: not enlarged (reassuring against malignancy)
- Significance
- Pathognomonic hydroxyapatite extrusion
- Sensitivity
- Low (most masses are intact)
- Specificity
- Very high
- Significance
- Distinguishes from solid tumour, lipoma, ganglion
- Sensitivity
- Moderate
- Specificity
- Moderate
- Significance
- Pattern recognition: 'cloud-like' or 'chicken-wire'
- Sensitivity
- High
- Specificity
- High in typical site
- Significance
- Suggests primary hyperphosphataemic type
- Sensitivity
- High in familial, low in secondary
- Specificity
- High for familial type
- Significance
- Suggests mature, organised deposit needing excision
- Sensitivity
- N/A
- Specificity
- N/A
Parosteal osteosarcoma arises from the periosteal surface, often of the posterior distal femur, with mature lamellar bone attached to the cortex and a 'string sign' (linear gap between tumour and cortex) on CT. Tumoral calcinosis is not attached to bone, is lobulated and amorphous (not bone-density), and does not invade cortex. When in doubt, biopsy before definitive surgery - and do not biopsy parosteal osteosarcoma through the planned resection tract.
- Typical Site
- Hip, shoulder, elbow, periarticular
- Key Feature
- Lobulated, periarticular, not attached to bone
- Discriminating Test
- X-ray pattern, serum phosphate, biopsy
- Typical Site
- Muscle belly (quadriceps, brachialis)
- Key Feature
- Mature lamellar bone, attached to periosteum over weeks
- Discriminating Test
- X-ray shows ossified rim with lucent centre; serial imaging
- Typical Site
- Posterior distal femur, proximal tibia, proximal humerus
- Key Feature
- Cortically based, mature bone, 'string sign'
- Discriminating Test
- CT, MRI, biopsy (only by sarcoma surgeon)
- Typical Site
- Intra-articular, knee, elbow, hip
- Key Feature
- Multiple intra-articular loose bodies, not periarticular mass
- Discriminating Test
- MRI, joint aspiration, arthroscopy
- Typical Site
- First MTP, ear, olecranon, Achilles
- Key Feature
- Soft tissue tophi, may calcify, hyperuricaemia
- Discriminating Test
- Serum urate, joint aspiration (urate crystals)
- Typical Site
- Fingertips, extensor surfaces
- Key Feature
- Small, multiple, skin-level calcifications
- Discriminating Test
- Clinical, ANA, capillaroscopy
- Typical Site
- Supraspinatus tendon
- Key Feature
- Hydroxylapatite within tendon, not lobulated mass
- Discriminating Test
- X-ray, ultrasound
- Typical Site
- Periarticular, vascular
- Key Feature
- Raised PTH, raised calcium in primary
- Discriminating Test
- Serum PTH, calcium, ultrasound of neck
The familial (hyperphosphataemic) form is a SYSTEMIC phosphate-handling disorder, so deposition is not confined to the periarticular soft tissue - and the extra-skeletal features are powerful diagnostic and counselling clues that examiners reward.
- Dental (near-characteristic): pulp stones, obliteration/narrowing of the pulp chambers, and short, bulbous ("thistle-shaped") roots - often visible incidentally on a panoramic dental radiograph and a useful confirmatory sign when the joint masses are atypical.
- Ocular: conjunctival and eyelid calcific deposits, and angioid streaks.
- Vascular: medial arterial calcification.
- Other: testicular microlithiasis has been reported.
Why it matters: a child or young adult with periarticular calcific masses plus the characteristic dental phenotype and hyperphosphataemia is hyperphosphataemic familial tumoral calcinosis until proven otherwise. These features support the diagnosis when imaging is equivocal, justify whole-patient surveillance, and inform genetic counselling for an autosomal recessive condition (relevant where consanguinity raises sibling risk).
TUMORALClinical Presentation of Tumoral Calcinosis
Hook:TUMORAL masses are not tumours - they are periarticular calcium hydroxyapatite deposits.
Investigations
Diagnostic Workup
Views: AP and lateral of the involved joint, plus a long-bone view
Look for: Lobulated, dense periarticular calcification; 'cloud-like' or 'chicken-wire' internal architecture; corticated or un-corticated margin; no bony attachment; no periosteal reaction
Sensitivity: High in typical site; can be diagnostic in familial or uraemic disease
Indication: Surgical planning, exclude bony attachment, confirm lobulated architecture
Look for: No continuity with cortex, no marrow involvement, no soft-tissue mass with calcified matrix suggesting sarcoma
Use: Differentiate from parosteal osteosarcoma, myositis ossificans, synovial chondromatosis
Indication: Atypical site, atypical imaging, diagnostic doubt
Look for: Homogeneous low T1 signal, variable T2 signal (often low because of calcium), no marrow oedema, no enhancing soft-tissue mass
Caution: Avoid biopsy until MRI and orthopaedic oncology review
Serum: Calcium, phosphate, magnesium, alkaline phosphatase, 25-OH vitamin D, 1,25-(OH)2 vitamin D, PTH, creatinine, eGFR, urate
Urine: 24-hour urinary phosphate (and calcium)
Calculations: Calcium-phosphate product, tubular reabsorption of phosphate (TRP), TmP/GFR
Aim: Separate primary hyperphosphataemic, primary normophosphataemic, secondary uraemic, hyperparathyroidism, vitamin D toxicity
Indications: Child or young adult, sibling cases, consanguinity, recurrence after excision, hyperphosphataemia with normal calcium and PTH
Genes: GALNT3, FGF23, KLOTHO, SLC34A3 (NaPi-IIc), SLC9A3R1 (NHERF1)
Method: Targeted panel or whole-exome sequencing with genetic counselling
Indication: Imaging atypical, mass enlarging rapidly, suspected malignancy, or first presentation in older patient without metabolic risk factors
Approach: Core needle biopsy by orthopaedic oncology service, placed in the planned resection tract
Histology: Lobulated basophilic calcium deposits with surrounding fibrosis, histiocytes and foreign-body giant cells; no atypia
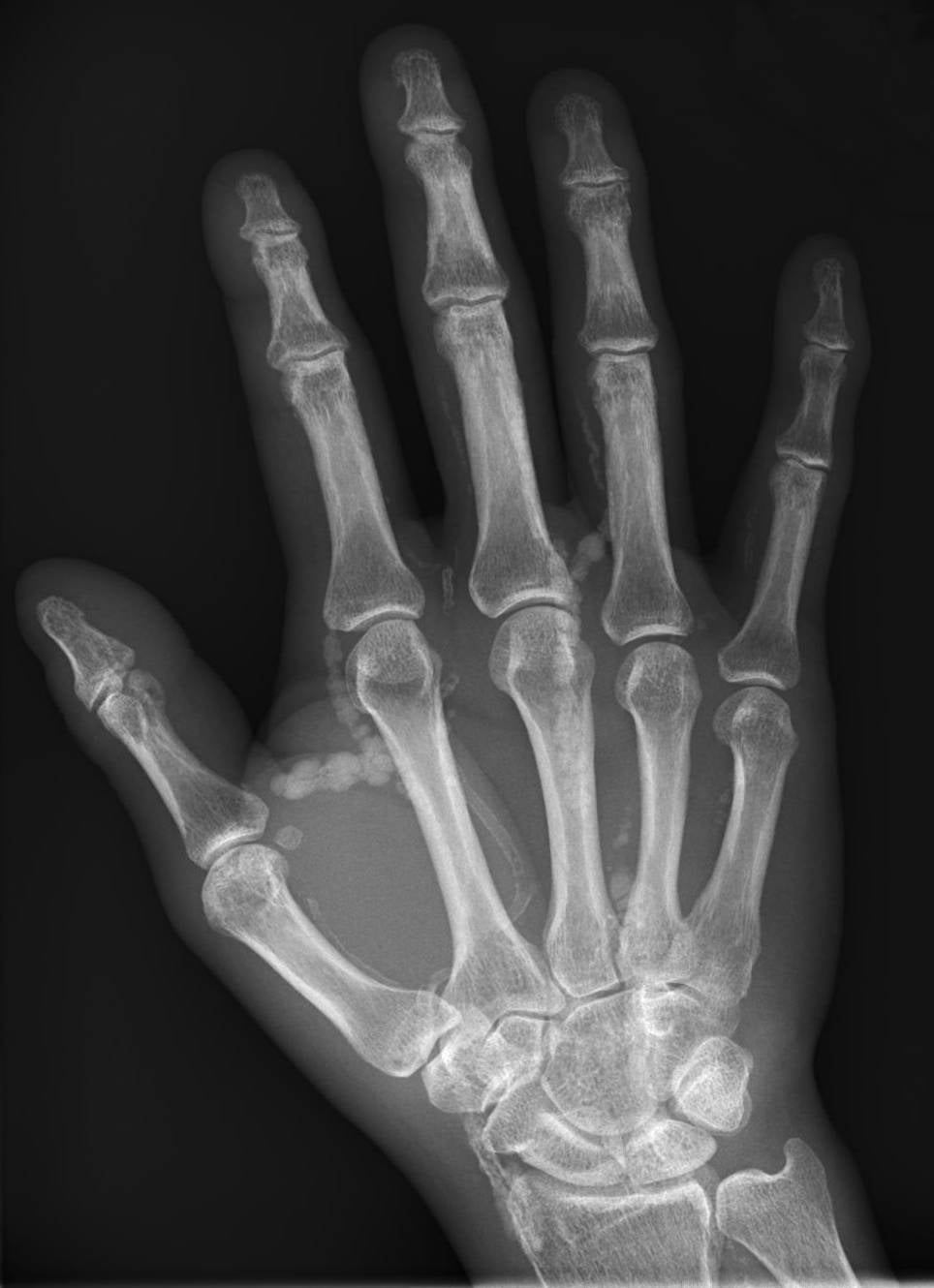
X-ray pattern recognition is often diagnostic. Lobulated, periarticular, 'cloud-like' calcification without cortical or medullary involvement in a young person = tumoral calcinosis until proven otherwise. CT or MRI is for surgical planning, not for diagnosis in the typical case. Always do the metabolic panel before any biopsy - the result may make the biopsy unnecessary.
- Primary Hyper-P
- Normal
- Primary Normo-P
- Normal
- Secondary (CKD)
- Low or normal (can be high with binders)
- Dystrophic / Other
- Normal (unless hyperparathyroidism)
- Primary Hyper-P
- Elevated (often greater than 2 mmol/L)
- Primary Normo-P
- Normal
- Secondary (CKD)
- Elevated
- Dystrophic / Other
- Variable
- Primary Hyper-P
- Normal or low
- Primary Normo-P
- Normal
- Secondary (CKD)
- Elevated (secondary hyperparathyroidism)
- Dystrophic / Other
- Variable
- Primary Hyper-P
- Low / suppressed
- Primary Normo-P
- Normal
- Secondary (CKD)
- Low (CKD)
- Dystrophic / Other
- Variable
- Primary Hyper-P
- Elevated (or inappropriately normal given high PO4)
- Primary Normo-P
- Normal
- Secondary (CKD)
- Markedly elevated
- Dystrophic / Other
- Normal
- Primary Hyper-P
- Normal
- Primary Normo-P
- Normal
- Secondary (CKD)
- Impaired (eGFR reduced or on dialysis)
- Dystrophic / Other
- Normal
Management Algorithm
Metabolic (Medical) Management - First Line
Goal: Lower serum phosphate, correct the calcium-phosphate product, and stop further deposition. This is the only durable therapy for familial disease and the only curative strategy for secondary uraemic disease.
Medical Protocol
Strict low-phosphate diet: avoid processed meats, cola drinks, dairy excess, nuts and beans
Typical target: dietary phosphate less than 800-1000 mg/day in adults
Dietician input: essential, ideally renal dietician for CKD patients
First-line (non-calcium): Sevelamer hydrochloride or sevelamer carbonate, lanthanum carbonate
Avoid calcium-based binders in patients with hyperphosphataemia and high Ca-P product because they worsen soft-tissue calcification
Dose: titrated to pre-dialysis or fasting serum phosphate
Mechanism: carbonic anhydrase inhibitor - increases renal phosphate excretion
Dose: 250-500 mg twice daily, monitor bicarbonate and potassium
Evidence: case series in familial tumoral calcinosis show mass reduction and phosphate fall
Caution: can worsen acidosis, hypokalaemia; not for advanced CKD
Mechanism: forms soluble calcium thiosulphate complexes
Route: topical (for ulcerated lesions) or intravenous (calciphylaxis-spectrum disease)
Evidence: anecdotal and small case series; not first-line
Hyperparathyroidism: parathyroidectomy, calcimimetics (cinacalcet)
Vitamin D toxicity: stop vitamin D analogues, calcium
Milk-alkali syndrome: stop intake, hydration
CKD-MBD: KDIGO-aligned bundle of diet, binders, dialysis adequacy, vitamin D analogues, calcimimetics
Acetazolamide is a useful adjunct in primary hyperphosphataemic disease because the kidneys are normal and can be coerced to waste more phosphate. It is not useful in uraemic secondary disease - the failing kidneys cannot respond. For uraemic disease, intensify dialysis, switch to non-calcium binders, and consider parathyroidectomy.
Complications
- Incidence
- Up to 30 percent in uraemic type, lower in familial
- Risk Factors
- Mass size, skin pressure, poor nutrition
- Management
- Wound care, infection control, complete excision when feasible
- Incidence
- 10-20 percent in ulcerated cases
- Risk Factors
- Skin breakdown, immunocompromise (dialysis)
- Management
- Antibiotics, surgical drainage, excision
- Incidence
- Variable, common with large masses
- Risk Factors
- Mass size, periarticular location
- Management
- Physiotherapy, surgical excision
- Incidence
- Uncommon but disabling
- Risk Factors
- Mass in deep anatomical site
- Management
- Urgent surgical decompression
- Incidence
- 50-100 percent in familial, lower in sporadic
- Risk Factors
- Incomplete excision, uncontrolled metabolism
- Management
- Metabolic control, complete re-excision, transplant if uraemic
- Incidence
- Up to 15 percent in ulcerated uraemic cases
- Risk Factors
- Poor tissue quality, uraemia, diabetes
- Management
- VAC therapy, flap reconstruction
- Incidence
- Rare, but high mortality
- Risk Factors
- Advanced CKD, warfarin, obesity
- Management
- Sodium thiosulphate, supportive care
Tumoral calcinosis is benign and biopsy is safe, but a presumed tumoral calcinosis that turns out to be a parosteal osteosarcoma must have its biopsy performed by the orthopaedic oncology service that will do the definitive resection. A poorly placed biopsy contaminates compartments and may cost the limb. If in doubt, refer for biopsy, do not perform it yourself.
Outcomes and Prognosis
- Medical Control
- Good with binders, diet, acetazolamide; new biologics emerging
- Surgical Cure Rate
- Low - high recurrence
- Long-Term Outlook
- Lifelong metabolic control; rare renal involvement
- Medical Control
- Limited - no metabolic driver to correct
- Surgical Cure Rate
- High if completely excised
- Long-Term Outlook
- Excellent if no recurrence; monitor for new lesions
- Medical Control
- Possible with aggressive CKD-MBD management
- Surgical Cure Rate
- Moderate - recurrence if metabolic state uncontrolled
- Long-Term Outlook
- Driven by renal disease; transplant is curative
- Medical Control
- Treat underlying cause
- Surgical Cure Rate
- Moderate to high
- Long-Term Outlook
- Cure if underlying cause controlled
Best prognosis: Sporadic normophosphataemic disease with complete primary excision; dystrophic disease with treated underlying cause.
Worst prognosis: Familial hyperphosphataemic disease with multiple recurrences; uraemic disease on long-term dialysis with poor metabolic control.
Curative treatment is achievable in two settings only: complete excision of a solitary normophosphataemic mass, and successful renal transplantation in uraemic disease.
The single biggest prognostic factor is whether the metabolic driver can be corrected - and in familial disease it usually cannot be cured, only controlled.
Guidelines, Registries & Global Practice
- Tumoral calcinosis is rare worldwide - incidence estimated at roughly 1 in 100,000 to 1 in 1,000,000
- Familial hyperphosphataemic form clusters in populations with high consanguinity - Middle East, North Africa, Turkey, parts of South Asia
- Secondary (uraemic) form is rising globally as the dialysis population grows - the most common clinical scenario in high-income settings
- Endemic clusters of tumoral calcinosis-like lesions have been described in sub-Saharan Africa and Papua New Guinea, often associated with low calcium, high phosphate dietary patterns
- Site distribution is consistent across regions: hip, shoulder, elbow, wrist, hand
- High-resource: tertiary orthopaedic oncology and metabolic bone units offer genetic testing, FGF23 assays, modern phosphate binders, calcimimetics, and access to renal transplantation
- Limited-resource: dietary phosphate restriction, low-cost aluminium-based or calcium-magnesium binders, and acetazolamide remain the backbone of therapy; genetic testing rarely available
- Dialysis adequacy is the strongest single modifiable factor in the uraemic form - more frequent or longer sessions are protective
- Surgical excision is widely available but recurrence remains high without metabolic correction
- No global arthroplasty or implant registry is relevant - tumoral calcinosis is a soft-tissue metabolic disease
- Diagnostic Emphasis
- Treat hyperphosphataemia and raised PTH first; binders, dialysis dose, calcimimetics, parathyroidectomy
- Medical Therapy
- Non-calcium binders first in high Ca-P product; cinacalcet for refractory secondary hyperparathyroidism
- Surgical Role
- Excision for symptomatic, ulcerating or infected masses; transplant is curative
- Diagnostic Emphasis
- Imaging pattern recognition (lobulated periarticular calcification) and biopsy only by sarcoma service when atypical
- Medical Therapy
- Treat underlying cause; refer to endocrinology for familial forms
- Surgical Role
- Complete primary excision preferred over debulking; recurrence is high in familial disease
- Diagnostic Emphasis
- Metabolic workup before any biopsy; X-ray pattern usually diagnostic
- Medical Therapy
- Phosphate binders, diet, acetazolamide; genetic testing for familial forms
- Surgical Role
- Excision reserved for symptomatic or diagnostic doubt cases
- Diagnostic Emphasis
- Recognise as rare disease; centralised reference centres for genetic and metabolic workup
- Medical Therapy
- Multidisciplinary metabolic bone service; emerging anti-FGF23 therapy
- Surgical Role
- Surgery adjunctive; conservative and metabolic management first
There is no dedicated registry for tumoral calcinosis, because it is a rare, non-implant, non-arthroplasty disease. The evidence base is dominated by small case series, mechanistic molecular studies, and narrative reviews. Familial disease is best registered through rare-disease networks (Orphanet, NIH GARD) and genotype-specific cohorts. The uraemic form is captured indirectly within CKD-MBD and dialysis registries.
Record in every patient with a periarticular calcific mass:
- Serum calcium, phosphate, PTH, 25-OH vitamin D, 1,25-(OH)2 vitamin D, creatinine, eGFR, urate, alkaline phosphatase
- Urinary phosphate and tubular reabsorption of phosphate
- Family history and consanguinity
- Renal history, dialysis vintage, current binders and vitamin D analogues
- Imaging: X-ray in two views, CT or MRI when atypical
- Biopsy tract (if performed) and the sarcoma service involvement when in doubt
- Plan for metabolic correction and, if needed, referral for genetic testing
A presumed 'tumour' that turns out to be tumoral calcinosis, or vice versa, is a recurring source of complaints worldwide. Always do the metabolic workup before any biopsy unless the imaging is unequivocally diagnostic and the biopsy is being done by the sarcoma service.
Controversies & Areas of Uncertainty
Familial hyperphosphataemic tumoral calcinosis almost always recurs after surgery, but a young patient with a large, ulcerating, infected mass cannot be left untreated. The threshold for surgery is therefore a clinical decision weighing symptoms, anatomy, recurrence risk, and the patient's wishes. There is no randomised evidence to guide this.
Burosumab is licensed for X-linked hypophosphataemia and tumour-induced osteomalacia, where it lowers phosphate by neutralising excess FGF23. In familial hyperphosphataemic tumoral calcinosis, FGF23 is elevated but dysfunctional - the response to burosumab is biologically plausible but unproven. Case reports suggest benefit, but there are no randomised trials, and cost and access are major barriers.
KDIGO suggests normalising phosphate in CKD-MBD, but in dialysis patients with established tumoral calcinosis the target is debated. Aggressive phosphate lowering may reduce new lesions but risks overshooting into hypophosphataemia, hungry bone, and adynamic bone disease. Individualised targets are accepted in practice but poorly evidence-based.
Complete primary excision is associated with lower recurrence than debulking, but in deep anatomical sites (sciatic notch, subdeltoid) complete excision carries real risk of nerve injury. There is no comparative trial. The pragmatic approach is: complete excision where safe, debulking where complete is unsafe, and aggressive metabolic control regardless.
Genetic testing is clearly useful in children, sibling cases, and consanguineous families. Its value in isolated adult cases, especially in uraemic patients, is less clear. Finding a heterozygous GALNT3 or FGF23 variant may not change management but does affect family counselling.
'Cure' means different things: anatomical (mass gone), biochemical (phosphate normal), genetic (mutation absent - impossible in familial), and patient-centred (symptom-free, functional). Most familial patients achieve only biochemical and patient-centred control, not anatomical cure.
MCQ Practice Points
Q: What is the molecular basis of autosomal recessive hyperphosphataemic familial tumoral calcinosis? A: Loss-of-function of GALNT3, FGF23 or KLOTHO, leading to impaired FGF23 signalling. GALNT3 glycosylates FGF23 so it can be secreted as an intact, biologically active phosphaturic hormone. Without intact FGF23 signalling, the kidney fails to waste phosphate, leading to hyperphosphataemia, suppressed 1,25-(OH)2 vitamin D, and progressive periarticular hydroxyapatite deposition.
Q: Classify tumoral calcinosis by pathogenesis. A: Using the Smack (1996) classification: primary (idiopathic) - further split into hyperphosphataemic familial (FGF23 axis) and normophosphataemic sporadic; secondary - chronic renal failure / dialysis, hyperparathyroidism, vitamin D toxicity, milk-alkali syndrome; dystrophic - local tissue injury; iatrogenic / miscellaneous - calcium-based binder excess, calcinosis cutis in connective tissue disease. The dominant clinical form is secondary (uraemic).
Q: What does the plain radiograph show in tumoral calcinosis, and how does it differ from parosteal osteosarcoma? A: Tumoral calcinosis shows a lobulated, periarticular, 'cloud-like' or 'chicken-wire' calcific mass that is not attached to the underlying bone. Parosteal osteosarcoma is a cortically based, mature-bone-forming mass with a 'string sign' on CT, often posterior distal femur, with potential marrow involvement. Biopsy of a suspected parosteal osteosarcoma must be performed by the sarcoma service.
Q: A young patient with periarticular calcific masses has normal calcium, phosphate of 2.6 mmol/L, suppressed 1,25-(OH)2 vitamin D and low-normal PTH. What is the most likely subtype? A: Primary hyperphosphataemic familial tumoral calcinosis. The biochemical pattern is high phosphate with normal calcium, low active vitamin D and low PTH. Most cases are due to biallelic GALNT3, FGF23 or KLOTHO mutations. Genetic testing is indicated.
Q: What is the first-line treatment for a patient with secondary (uraemic) tumoral calcinosis on dialysis? A: Metabolic correction, not surgery. Switch from calcium-based to non-calcium phosphate binders (sevelamer, lanthanum), intensify dialysis, review vitamin D analogue dose, target pre-dialysis phosphate less than 1.5 mmol/L, and consider parathyroidectomy if PTH remains very high. Surgical excision is reserved for symptomatic (ulcerating, infected, mechanically limiting) masses. Renal transplantation is the only curative metabolic therapy.
Q: How do you distinguish tumoral calcinosis from myositis ossificans? A: Tumoral calcinosis is a periarticular, lobulated, calcium hydroxyapatite mass, typically around the hip, shoulder or elbow, in a patient with a metabolic driver (familial FGF23 axis defect or chronic renal failure). Myositis ossificans is a mature lamellar bone mass within a muscle belly (commonly quadriceps or brachialis), typically after trauma, attached to periosteum, with a peripheral ossified rim and a lucent centre on serial imaging. Tumoral calcinosis does not form bone; myositis ossificans does.
Q: What is the recurrence rate after surgical excision of familial hyperphosphataemic tumoral calcinosis? A: High, often greater than 50 percent, sometimes approaching 100 percent after simple debulking. Recurrence is the rule because surgery does not correct the underlying FGF23 axis defect. The only curative approach is metabolic control (phosphate binders, acetazolamide, low-phosphate diet, emerging anti-FGF23 antibodies) and, for solitary normophosphataemic masses, complete primary excision.
Exam Viva Scenarios
Practise clinical reasoning and management decisions out loud
“A 58-year-old man on haemodialysis for 9 years (end-stage renal disease from diabetes) presents with a slowly enlarging, firm, lobulated mass over the greater trochanter of his right hip. It is non-tender, stony-hard, and the overlying skin is intact. He is on calcium carbonate as a phosphate binder and alfacalcidol. Serum calcium is 2.4 mmol/L, phosphate is 2.6 mmol/L, PTH is 850 pg/mL, and 25-OH vitamin D is 30 nmol/L. What is the diagnosis and how do you manage him?”
“A 14-year-old boy, born to consanguineous parents, presents with recurrent lobulated masses around both hips and his right elbow. He had his first mass excised at age 8 with recurrence within a year. Serum calcium is normal, phosphate is 2.7 mmol/L, PTH is low-normal, 1,25-(OH)2 vitamin D is low, FGF23 is inappropriately normal for the degree of hyperphosphataemia, and renal function is normal. Genetic testing reveals homozygous GALNT3 mutations. How do you counsel and manage him?”
Definition and Pathology
- Periarticular, lobulated deposit of calcium hydroxyapatite - not a true neoplasm
- Mineral is calcium phosphate (hydroxyapatite), not calcium oxalate or CPPD
- Lobulated architecture with foreign-body giant cell reaction; no cellular atypia
- Most common sites: hip, shoulder, elbow, wrist
Classification (Smack 1996)
- Primary hyperphosphataemic (familial) - GALNT3, FGF23, KLOTHO mutations
- Primary normophosphataemic (sporadic) - often solitary
- Secondary (uraemic) - chronic renal failure, dialysis, high Ca-P product
- Dystrophic - local trauma, hyperparathyroidism, vitamin D toxicity, milk-alkali
Diagnosis
- X-ray: lobulated, 'cloud-like' or 'chicken-wire' periarticular calcification, not attached to bone
- CT: confirms no cortical continuity; MRI for atypical cases or surgical planning
- Lab panel: calcium, phosphate, PTH, 25-OH vitamin D, 1,25-(OH)2 vitamin D, creatinine, FGF23, urinary phosphate
- Biopsy only by sarcoma service when imaging atypical; metabolic workup before biopsy
FGF23 Axis
- FGF23 is secreted by osteocytes; requires GALNT3 O-glycosylation for intact secretion
- Binds FGFR1-KLOTHO complex in the kidney to promote phosphate wasting and suppress 1-alpha hydroxylase
- Loss-of-function of GALNT3, FGF23 or KLOTHO = hyperphosphataemia, suppressed 1,25-(OH)2 vitamin D
- Net effect: high phosphate, normal calcium, normal or low PTH, periarticular hydroxyapatite
Differential Diagnosis
- Myositis ossificans - mature lamellar bone in muscle, attached to periosteum, post-trauma
- Parosteal osteosarcoma - cortically based, mature bone, 'string sign', biopsy only by sarcoma service
- Synovial osteochondromatosis - intra-articular loose bodies, not periarticular mass
- Tophaceous gout - urate tophi, hyperuricaemia, joint aspiration with urate crystals
Medical Management
- Low-phosphate diet (target less than 800-1000 mg/day in adults)
- Non-calcium phosphate binders (sevelamer, lanthanum) - avoid calcium binders with high Ca-P product
- Acetazolamide 250-500 mg twice daily - useful in familial, not in uraemic
- Treat underlying cause: parathyroidectomy, stop vitamin D analogues, transplant in CKD
Surgical Management
- Indications: ulceration, infection, nerve compression, mechanical block, diagnostic doubt, cosmesis
- Technique: complete primary extracapsular excision preferred over debulking
- Recurrence rate: 50-100 percent in familial disease, lower in sporadic
- Re-excision only for genuinely symptomatic recurrence after metabolic optimisation
Prognosis and Pitfalls
- Familial disease is controlled, not cured - lifelong phosphate management required
- Secondary (uraemic) disease can be cured by successful renal transplantation
- Sporadic normophosphataemic disease has the best prognosis after complete excision
- Do not biopsy a suspected parosteal osteosarcoma through the wrong tract - refer to sarcoma service
Evidence Base and Key Trials
Proposal for a pathogenesis-based classification of tumoral calcinosis
- Classifies tumoral calcinosis into primary (normophosphataemic and hyperphosphataemic familial) and secondary (uraemic, hyperparathyroidism, vitamin D toxicity, milk-alkali, connective tissue disease) subtypes based on pathogenesis rather than site
- Hyperphosphataemic familial subtype clusters in children of consanguineous parents, with autosomal recessive inheritance and high post-excision recurrence
- Secondary subtype is far more common clinically because of the dialysis population, with calcium-phosphate product as the principal driver
- Treatment is metabolic first in secondary disease, surgical only for symptomatic masses
Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis
- Identified biallelic loss-of-function mutations in GALNT3 as the cause of autosomal recessive hyperphosphataemic familial tumoral calcinosis
- GALNT3 encodes a glycosyltransferase required for O-linked glycosylation and secretion of intact, biologically active FGF23
- Affected patients had elevated serum phosphate, normal calcium, suppressed 1,25-(OH)2 vitamin D, normal or low PTH, and active FGF23 substrate accumulation
- Established the FGF23-GALNT3-KLOTHO axis as central to phosphate handling
A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis
- Identified biallelic FGF23 mutations in a family with autosomal recessive hyperphosphataemic tumoral calcinosis, complementing GALNT3 findings
- FGF23 mutations impair either FGF23 protein stability or receptor binding, leading to loss of phosphaturic signalling
- Patients had the classic biochemical triad - high phosphate, normal calcium, suppressed 1,25-(OH)2 vitamin D
- Demonstrated locus heterogeneity in familial tumoral calcinosis
Phenotypic and genotypic characterization and treatment of a cohort with familial tumoral calcinosis/hyperostosis-hyperphosphatemia syndrome
- Multicentre NIH-led cohort of 8 patients with familial tumoral calcinosis/hyperostosis-hyperphosphatemia syndrome, all with hyperphosphataemia, increased tubular reabsorption of phosphate, and elevated or inappropriately normal 1,25-(OH)2 vitamin D
- Biallelic GALNT3 mutations identified in 7 of 8 subjects; FGF23 and KLOTHO recognised as the other causal genes in the FTC/HHS spectrum
- Clinical phenotype ranged from asymptomatic to massive, disabling calcifications, with diaphyseal hyperostosis on radiographs; one subject had complete resolution of a calcific mass after 13 months of medical therapy
- Two subjects with severe calcifications also had overwhelming systemic inflammation that responded to IL-1 antagonists, a novel finding in FTC/HHS
Review of tumoral calcinosis: a rare clinico-pathological entity
- Clinicopathological review of tumoral calcinosis confirming the primary (hyperphosphataemic familial and normophosphataemic sporadic) and secondary (chronic renal failure, hyperparathyroidism) subtypes
- Hyperphosphataemic familial form attributed to mutations in GALNT3, KLOTHO or FGF23; normophosphataemic primary form increasingly linked to SAMD9 variants
- Endorses a stage-oriented approach: phosphate binders, low-phosphate diet and acetazolamide should be tried before surgery in primary disease because of high recurrence and complication rates
- Medical therapy is the mainstay for the secondary uraemic variety, with parathyroidectomy reserved for failure; surgical excision in uraemic patients is a last resort