Low Alkaline Phosphatase and Impaired Mineralization
- Hypophosphatasia (HPP) is an inherited metabolic bone disease caused by LOSS-OF-FUNCTION mutations in the ALPL gene, which encodes TISSUE-NONSPECIFIC ALKALINE PHOSPHATASE (TNSALP); the enzyme deficiency impairs skeletal and dental mineralization.
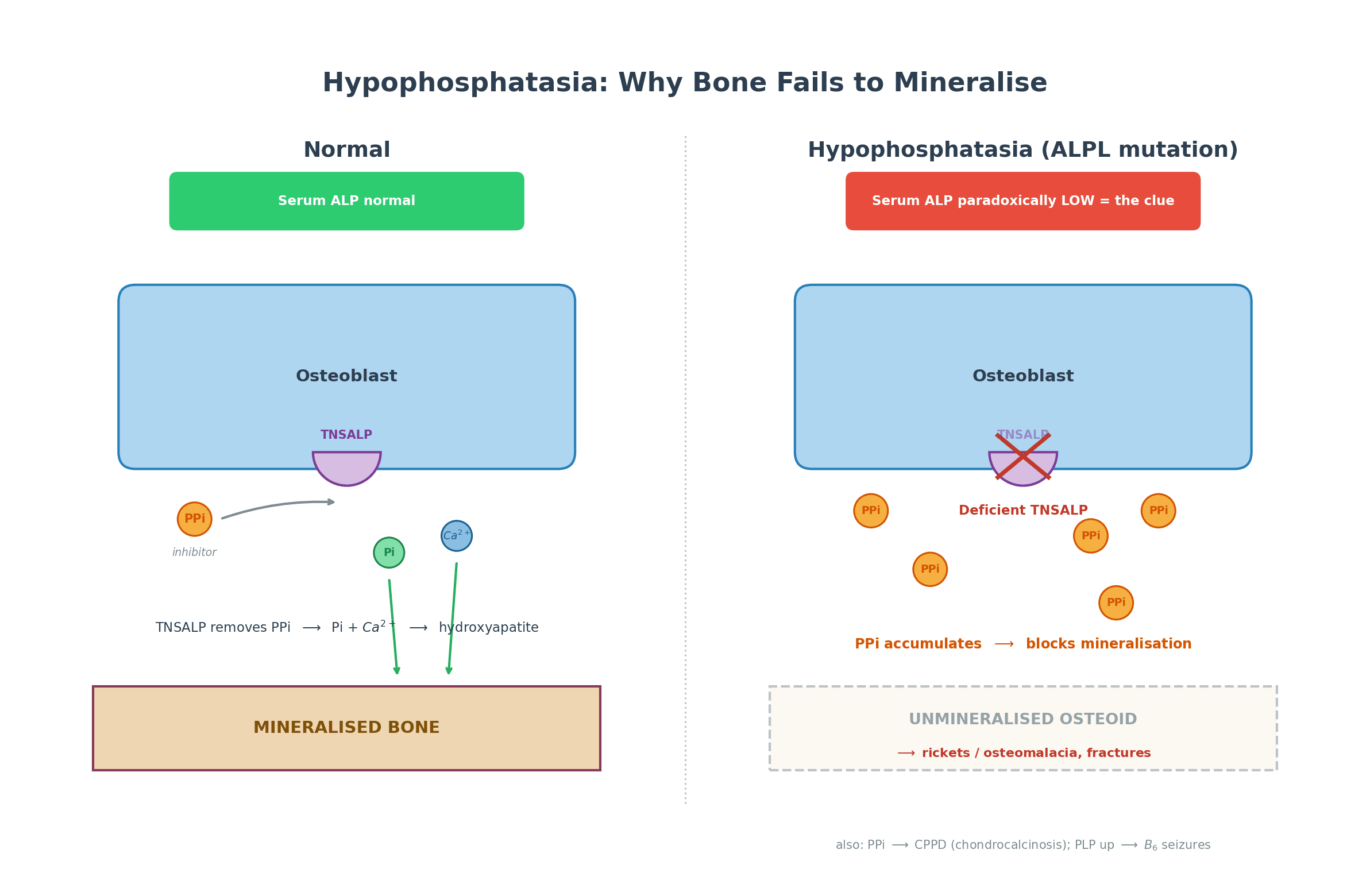
- The DIAGNOSTIC CLUE is a paradoxically LOW serum ALKALINE PHOSPHATASE - the opposite of most bone diseases (rickets, osteomalacia, Paget's, fracture healing) which RAISE ALP - so a persistently LOW ALP in a patient with fractures or bone disease should prompt the diagnosis.
- Deficient TNSALP causes accumulation of its substrates: INORGANIC PYROPHOSPHATE (PPi - a potent inhibitor of mineralization, so it builds up and blocks bone formation and causes chondrocalcinosis/pseudogout), PYRIDOXAL-5'-PHOSPHATE (PLP/vitamin B6 - hence B6-dependent seizures in severe infantile disease), and PHOSPHOETHANOLAMINE (PEA, raised in urine).
- The clinical SPECTRUM is broad and severity is inversely related to age of onset: perinatal (often lethal) and infantile (failure to thrive, rickets, HYPERCALCAEMIA, craniosynostosis, seizures), childhood (rickets, bowing, short stature, and the characteristic PREMATURE LOSS OF PRIMARY TEETH with intact roots), adult (osteomalacia, recurrent poorly-healing stress/pseudofractures of the metatarsals and subtrochanteric femur, chondrocalcinosis/pseudogout), and odonto-HPP (dental only).
- The cardinal ORTHOPAEDIC PITFALL: BISPHOSPHONATES are CONTRAINDICATED in HPP - they are pyrophosphate analogues that further inhibit mineralization and can precipitate ATYPICAL FEMORAL FRACTURES; a low ALP must be recognised BEFORE prescribing them for a 'fragility' fracture.
- DIAGNOSIS combines a low ALP with raised substrates (serum PLP/B6, urinary PEA), characteristic radiographs (rickets with 'tongue-like' metaphyseal lucencies in children; osteomalacia/pseudofractures in adults) and ALPL genetic testing; the key TREATMENT advance is ASFOTASE ALFA, a bone-targeted recombinant TNSALP enzyme-replacement therapy that transforms outcomes in perinatal/infantile/childhood HPP, with supportive care, B6 for seizures, and avoidance of bisphosphonates and excess vitamin D.
- “HPP = ALPL/TNSALP deficiency; the clue is a LOW serum alkaline phosphatase (opposite of other bone diseases).
- “Substrate build-up: PPi (inhibits mineralization, chondrocalcinosis), PLP/B6 (seizures), urinary PEA. Childhood hallmark = premature loss of PRIMARY teeth.
- “Do NOT give bisphosphonates (PPi analogues - worsen it / atypical femoral fractures). Treat with asfotase alfa (enzyme replacement).
A persistently LOW serum ALP with fractures/bone disease, premature tooth loss, or chondrocalcinosis = think hypophosphatasia (most bone diseases RAISE ALP).
Do NOT give bisphosphonates (pyrophosphate analogues) - they further inhibit mineralization and risk atypical femoral fractures. Treat with asfotase alfa.
Biochemistry & Pathophysiology
TNSALP normally hydrolyses inorganic pyrophosphate (PPi) at the mineralizing surface; PPi is a potent inhibitor of hydroxyapatite formation, so removing it allows mineralization. In HPP, deficient TNSALP lets PPi accumulate, which blocks mineralization (rickets/osteomalacia) and deposits as calcium pyrophosphate (chondrocalcinosis/pseudogout). The enzyme's other substrates also build up: pyridoxal- 5'-phosphate (PLP, vitamin B6) - its failure to enter the brain causes the B6-dependent seizures of severe infantile disease - and phosphoethanolamine (PEA), raised in the urine. Crucially, because the enzyme itself is deficient, serum alkaline phosphatase is LOW - the diagnostic signature.
Clinical Spectrum & Diagnosis
Severity is inversely related to age of onset:
- Perinatal: profound hypomineralization, frequently lethal (respiratory failure).
- Infantile: failure to thrive, rickets, hypercalcaemia/hypercalciuria, craniosynostosis, seizures, respiratory problems.
- Childhood: rickets, bowing, short stature, delayed/waddling gait, and the characteristic premature loss of primary teeth with the root intact.
- Adult: osteomalacia, recurrent, poorly-healing stress/pseudofractures (classically metatarsals and the subtrochanteric femur), chondrocalcinosis/pseudogout, early tooth loss and chronic musculoskeletal pain.
- Odonto-HPP: dental disease only. Diagnosis: a LOW serum ALP (age/sex-adjusted) plus raised substrates (serum PLP/B6, urinary PEA), characteristic radiographs ('tongue-like' metaphyseal radiolucencies in children; osteomalacia/ Looser-zone pseudofractures in adults), and ALPL genetic testing.
A Low ALP Is Not Always HPP: The Differential
The whole topic turns on "recognise the low ALP" and warns against "confusing it with rickets" - so know both sides of the differential:
- HPP versus the rachitic/osteomalacic mimics - use the ALP direction: nutritional (vitamin-D-deficiency) rickets, renal osteodystrophy and hypophosphataemic rickets/osteomalacia (XLH, oncogenic) all RAISE the ALP as osteoblasts work overtime, whereas HPP LOWERS it - so a child with a rachitic radiograph but a low ALP has HPP until proven otherwise. Calcium/phosphate help too (HPP infants are often hypercalcaemic, unlike the low/normal calcium of nutritional rickets). (The mimics themselves are in our Rickets, Vitamin D Deficiency and XLH/Oncogenic Osteomalacia topics.)
- The other causes of a genuinely low ALP - exclude before diagnosing HPP: because ALP is a zinc/magnesium metalloenzyme and a marker of bone/liver turnover, it falls in hypothyroidism, hypomagnesaemia/zinc deficiency, malnutrition, severe anaemia, Wilson's disease, glucocorticoid excess/Cushing's, recent bisphosphonate or denosumab therapy, vitamin D toxicity, multiple myeloma, massive transfusion and major illness. So repeat an age- and sex-adjusted ALP, exclude these, and only then pursue HPP with the raised substrates (PLP/B6, urinary PEA) and ALPL genetics.
A low ALP narrows to HPP only after two steps: (1) distinguish HPP (LOW ALP) from the rickets/osteomalacia mimics (which raise ALP); (2) exclude the other causes of a low ALP - hypothyroidism, hypomagnesaemia/zinc deficiency, malnutrition, Wilson's, steroids, antiresorptive therapy, myeloma. Then confirm with raised substrates + ALPL genetics.
Management
- Asfotase alfa: a bone-targeted recombinant TNSALP enzyme-replacement therapy that markedly improves survival, skeletal mineralization and growth in perinatal/infantile/childhood HPP - early diagnosis is essential to start it in time.
- Supportive/multidisciplinary: vitamin B6 for the B6-dependent seizures of severe infantile disease; respiratory support; orthopaedic care of fractures and deformity; dental care; physiotherapy; and management of hypercalcaemia in infants.
- AVOID: bisphosphonates (pyrophosphate analogues - worsen the defect and risk atypical femoral fractures) and excessive vitamin D/calcium loading (hypercalcaemia); teriparatide has been used off-label in some adults.
- Fracture care: fractures heal poorly, so optimise fixation, anticipate delayed/non-union, and treat the underlying disease (asfotase alfa where indicated).
The single most important practical message is that a patient presenting with fragility or stress fractures and a persistently LOW alkaline phosphatase may have hypophosphatasia, in which bisphosphonates are contraindicated - giving them is a recognised error that can worsen mineralization and precipitate atypical femoral fractures. Check the ALP before treating a 'fragility fracture' as routine osteoporosis, and refer suspected HPP to metabolic-bone specialists for confirmation and enzyme-replacement consideration.
Inheritance: Why Some Are Recessive and Some Dominant
HPP arises from loss-of-function ALPL mutations, and the compound heterozygous versus heterozygous split in the cited series turns on the inheritance pattern itself - high-yield for counselling:
- The gene: ALPL (on chromosome 1) encodes TNSALP, which works as a functional dimer; there are many disease-causing variants, giving the broad genotype-phenotype spectrum.
- Severe early-onset disease is autosomal RECESSIVE: the perinatal and infantile forms require two loss-of-function alleles (homozygous or compound heterozygous).
- Milder disease can be autosomal DOMINANT or recessive: the childhood, adult and odonto forms may arise from a single allele - particularly a dominant-negative variant, where the mutant subunit poisons the wild-type partner in the dimer. This is why a parent or sibling with only a low ALP (a carrier, or mild/odonto HPP) may be found when a severely affected child is diagnosed.
- Implication: the recurrence risk and counselling depend on the pattern - biallelic for severe disease (each parent a carrier), versus a dominant transmission for some milder pedigrees. Test the family, not just the index patient (general principles in our Genetics & Inheritance in Orthopaedics topic).
Severe HPP (perinatal/infantile) = autosomal recessive (two loss-of-function ALPL alleles); milder (childhood/adult/odonto) can be dominant or recessive, often via a dominant-negative variant that poisons the TNSALP dimer. So an asymptomatic relative with an isolated low ALP may be a carrier or have mild HPP - test the family.
Mnemonics & Memory Aids
LOW ALP
Hook:Everything turns on a LOW ALP.
DON'T GIVE
Hook:In HPP, DON'T GIVE bisphosphonates - GIVE asfotase alfa.
Clinical Decision Scenarios
Practise clinical reasoning and management decisions out loud
“An adult presents with recurrent metatarsal stress fractures and a persistently low alkaline phosphatase. What diagnosis must you consider, and why does it change management?”
“How does hypophosphatasia present across the age spectrum, and what is the diagnostic biochemistry?”
Cause & biochemistry
- ALPL loss-of-function -> deficient TNSALP
- LOW serum alkaline phosphatase (the clue; others RAISE ALP)
- Substrate build-up: PPi (inhibits mineralization), PLP/B6 (seizures), urinary PEA
Spectrum
- Perinatal (lethal) / infantile (FTT, hypercalcaemia, seizures)
- Childhood: rickets + premature loss of PRIMARY teeth (intact roots)
- Adult: osteomalacia, metatarsal/subtrochanteric pseudofractures, chondrocalcinosis; odonto = dental only
Diagnosis
- Low ALP + raised serum PLP (B6) + urinary PEA
- Radiographs: tongue-like metaphyseal lucencies (child) / Looser zones (adult)
- ALPL genetic testing
Management
- Asfotase alfa (recombinant TNSALP ERT) - early diagnosis essential
- B6 for seizures; treat fractures (poor healing); MDT/dental care
- AVOID bisphosphonates (atypical femoral fractures) and excess vitamin D
Evidence & Key Studies
Hypophosphatasia in childhood: diagnosis to management
- HPP results from loss-of-function ALPL mutations causing deficient TNSALP activity and is diagnosed from combined clinical, laboratory (low ALP), radiographic and genetic findings.
- Clinical heterogeneity makes diagnosis challenging, so early recognition is essential.
- Asfotase alfa, a bone-targeted recombinant TNSALP enzyme-replacement therapy, has improved prognosis.
Phenotypic and genotypic spectrum of Indian patients with hypophosphatasia
- Five genetically proven HPP patients ranged from an infant (failure to thrive, hypercalcaemia, low ALP) and an adolescent (genu valgum, delayed dentition, 'tongue-like' metaphyseal translucencies) to adults with vertebral fractures and musculoskeletal pain.
- Compound heterozygous ALPL variants caused severe early disease; heterozygous variants caused milder adult disease; an infant inappropriately received bisphosphonate before HPP was recognised.
- Asfotase alfa produced remarkable growth improvement, underscoring the need to recognise a low ALP and avoid harmful treatment.
The ALPL/TNSALP basis, the diagnostic combination including a low ALP, and the role of asfotase alfa come from the cited Im & Cho review, and the clinical spectrum (infantile hypercalcaemia, 'tongue-like' metaphyseal translucencies, adult fractures), the harm of inappropriate bisphosphonate use and the response to asfotase alfa from the cited Dhananjaya case series. The substrate biochemistry (PPi, PLP/B6, PEA) and the bisphosphonate contraindication are standard, well-established teaching. (See also our Rickets/ Osteomalacia and Calcium/Metabolic Bone topics.)