Benign Bone-Forming Tumor | Posterior Elements | Pain Syndrome
Enneking Classification (Benign)
Critical Must-Knows
- Osteoblastoma is histologically similar to osteoid osteoma but larger than 2cm and lacks nidus
- About 30-40% occur in spine, particularly posterior elements (pedicle, lamina, transverse process)
- Pain is NOT typically relieved by NSAIDs (key distinguishing feature from osteoid osteoma)
- CT shows expansile lytic lesion with variable mineralization and 'mini-brain' appearance
- Treatment is intralesional curettage with adjuvants or en bloc resection depending on stage
Clinical Pearls
- "Distinguish from osteoid osteoma: larger than 2cm, spine location, NSAIDs ineffective
- "Aggressive osteoblastoma can mimic osteosarcoma - requires expert histopathology review
- "Recurrence rate 10-20% after intralesional treatment, higher in aggressive variant
- "Spinal lesions may present with neurological deficit from mass effect or pathological fracture
Clinical Imaging
Imaging Gallery




Critical Osteoblastoma Exam Points
Histological Confusion
Aggressive osteoblastoma mimics osteosarcoma. Requires expert histopathology review. Key differences: circumscribed margin, uniform cellularity, no permeative growth pattern.
Differential Diagnosis
Distinguish from osteoid osteoma: Osteoblastoma larger than 2cm, lacks discrete nidus, NSAIDs ineffective for pain. Both produce osteoid but different clinical behavior.
Spinal Predilection
About 30-40% occur in spine (posterior elements). Can cause neurological deficit, scoliosis, or pathological fracture. CT-guided biopsy essential before surgery.
Treatment Principles
Intralesional excision with adjuvants (phenol, PMMA) for Stage 2. En bloc resection for aggressive variant. Recurrence 10-20%. Spine requires decompression and stabilization.
At a Glance
Osteoblastoma is a rare benign bone-forming tumor comprising 1% of primary bone tumors, with peak incidence in patients aged 10-30 years and male predominance (2:1). The critical distinguishing feature from osteoid osteoma is size greater than 2cm, lack of discrete nidus, and importantly, pain NOT relieved by NSAIDs. Approximately 30-40% involve the spine (32% in the 306-case Lucas series), particularly posterior elements (pedicle, lamina), where neurological deficit or scoliosis may develop. Imaging reveals an expansile lytic lesion with variable mineralization and characteristic "mini-brain appearance" on CT. Treatment follows Enneking staging: intralesional curettage with adjuvants for Stage 2 lesions, with en bloc resection for aggressive variants that can histologically mimic osteosarcoma—recurrence rates are 10-20% after intralesional treatment.
BLASTOsteoblastoma Key Features
| B | Benign bone-forming tumor Produces osteoid and woven bone |
| L | Larger than 2cm Key size criterion distinguishing from osteoid osteoma |
| A | Aspirin ineffective Pain NOT relieved by NSAIDs (unlike osteoid osteoma) |
| S | Spine posterior elements 30-40% spinal, especially pedicle and lamina |
| T | Teens to young adults Peak incidence 10-30 years, male greater than female 2:1 |
| B | Benign bone-forming tumor Produces osteoid and woven bone | S | Spine posterior elements 30-40% spinal, especially pedicle and lamina |
| L | Larger than 2cm Key size criterion distinguishing from osteoid osteoma | T | Teens to young adults Peak incidence 10-30 years, male greater than female 2:1 |
| A | Aspirin ineffective Pain NOT relieved by NSAIDs (unlike osteoid osteoma) |
Hook:BLAST = Big Lesion with Aspirin-resistant pain in Spine of Teens!
MINEImaging Characteristics
| M | Mini-brain appearance Characteristic CT finding with lace-like trabeculae |
| I | Intramedullary location Usually metaphysis or diaphysis, less common in epiphysis |
| N | Nidus absent Unlike osteoid osteoma which has discrete nidus |
| E | Expansile lytic lesion Variable mineralization, may have thin sclerotic rim |
| M | Mini-brain appearance Characteristic CT finding with lace-like trabeculae | N | Nidus absent Unlike osteoid osteoma which has discrete nidus |
| I | Intramedullary location Usually metaphysis or diaphysis, less common in epiphysis | E | Expansile lytic lesion Variable mineralization, may have thin sclerotic rim |
Hook:MINE the characteristics: Mini-brain, Intramedullary, No nidus, Expansile!
OABCDifferential Diagnosis
| O | Osteoid osteoma Smaller than 2cm, nidus, aspirin-responsive pain |
| A | Aneurysmal bone cyst Fluid-fluid levels on MRI, no osteoid production |
| B | Bone island (enostosis) Dense sclerotic, no expansion, asymptomatic |
| C | Chondroblastoma Epiphyseal location, chondroid matrix, younger age |
| O | Osteoid osteoma Smaller than 2cm, nidus, aspirin-responsive pain | B | Bone island (enostosis) Dense sclerotic, no expansion, asymptomatic |
| A | Aneurysmal bone cyst Fluid-fluid levels on MRI, no osteoid production | C | Chondroblastoma Epiphyseal location, chondroid matrix, younger age |
Hook:OABC differential: Osteoid osteoma, ABC, Bone island, Chondroblastoma!
Overview and Epidemiology
Osteoblastoma is a rare benign bone-forming tumor characterized by production of osteoid and immature woven bone by active osteoblasts. It represents approximately 1% of all primary bone tumors and 3% of benign bone tumors. The tumor is histologically similar to osteoid osteoma but distinguished by size greater than 2cm and different clinical behavior.
Clinical Significance
Osteoblastoma is clinically important because: (1) it can mimic malignant tumors radiologically and histologically, particularly the aggressive variant; (2) spinal involvement may cause neurological compromise; (3) local recurrence occurs in 10-20% requiring long-term surveillance; and (4) distinguishing from osteoid osteoma is essential as treatment approaches differ.
Demographics
- Age: Peak 10-30 years (range 2-75 years)
- Sex: Male greater than female 2:1
- Location: 30-40% spine, ~30% long bones, remainder other sites
- Ethnic: No racial predilection
Anatomical Distribution
- Spine: Posterior elements (pedicle, lamina, transverse process)
- Long bones: Femur, tibia (metaphysis or diaphysis)
- Other: Mandible, hands, feet (less common)
- Epiphysis: Rare (under 5% of cases)
Pathophysiology and Histology
Pathogenesis
Osteoblastoma arises from osteoblastic cells that produce osteoid and woven bone. The defining molecular event is now established: recurrent rearrangements of FOS, and less commonly FOSB, are present in the great majority of osteoblastomas and osteoid osteomas, placing the two lesions in a single molecular family. These rearrangements are absent in osteosarcoma, so FOS immunohistochemistry (or FOS/FOSB FISH) is a useful ancillary test when morphology alone cannot separate an aggressive osteoblastoma from a bone-forming sarcoma.
Aggressive Osteoblastoma Variant
Approximately 10-15% of osteoblastomas demonstrate aggressive histological features including epithelioid osteoblasts, sheet-like growth, and increased mitotic activity. This aggressive variant has higher recurrence rate (25-50%) and can be misdiagnosed as osteosarcoma. Expert musculoskeletal pathology review is mandatory.
Histological Features
Histological Characteristics
| Feature | Classic Osteoblastoma | Aggressive Variant | Osteosarcoma |
|---|---|---|---|
| Growth pattern | Circumscribed, pushing margin | Infiltrative at periphery | Permeative, destructive |
| Osteoblasts | Uniform, single nuclei | Epithelioid, prominent nucleoli | Atypical, pleomorphic |
| Osteoid production | Abundant, lace-like | Variable, irregular | Minimal to abundant, atypical |
| Mitotic activity | Rare (under 1 per HPF) | Increased (2-5 per HPF) | Frequent (over 10 per HPF) |
| Vascular stroma | Prominent, dilated vessels | Variable vascularity | Often necrotic areas |
Microscopic Features
- Osteoid: Abundant production of immature osteoid
- Bone: Woven bone with irregular mineralization
- Osteoblasts: Uniform cells lining osteoid seams
- Stroma: Highly vascular fibrous tissue
- Giant cells: Scattered osteoclast-like giant cells
Key Distinguishing Features
- vs Osteoid osteoma: Larger size, no discrete nidus
- vs Osteosarcoma: Circumscribed margin, uniform cells, no permeation
- vs ABC: Produces osteoid, no fluid-fluid levels
- vs GCT: Younger age, osteoid production present
Clinical Presentation
Symptoms
The classic presentation is progressive bone pain over weeks to months, often worse at night. Unlike osteoid osteoma, the pain is typically NOT relieved by aspirin or NSAIDs (important distinguishing feature). Spinal lesions may present with radicular pain, motor weakness, or scoliosis.
Pain Characteristics
- Duration: Progressive over weeks to months
- Pattern: Constant, worse at night
- Severity: Moderate to severe
- NSAID response: Ineffective (key difference from osteoid osteoma)
- Radiation: May have radicular component if spinal
Neurological Features (Spinal)
- Radiculopathy: Nerve root compression
- Motor deficit: Weakness in myotomal distribution
- Sensory deficit: Dermatomal numbness
- Scoliosis: Painful scoliosis in children
- Cauda equina: Rare with lower lumbar lesions
Physical Examination
Systematic Examination Approach
- Swelling: Visible if superficial (hands, feet)
- Deformity: Scoliosis with spinal lesions
- Gait: Antalgic gait if lower limb involved
- Skin: Normal overlying skin (no warmth or redness)
- Tenderness: Localized bony tenderness
- Mass: Palpable if cortical expansion
- Temperature: Normal (not warm)
- Neurovascular: Check distal pulses and sensation
- ROM: Limited by pain if juxta-articular
- Strength: Reduced if pathological fracture or nerve compression
- Neurological: Full examination if spinal (motor, sensory, reflexes)
- Spine: Straight leg raise, neurological level
- Pathological fracture: Stability assessment
- Scoliosis: Adams forward bend test
Red Flags Requiring Urgent Assessment
Immediate evaluation needed if:
- Progressive neurological deficit (spinal cord or cauda equina compression)
- Severe unremitting pain suggesting pathological fracture
- Systemic symptoms (fever, weight loss) suggesting infection or malignancy
- Rapid progression of symptoms over days to weeks
Investigations and Imaging
Plain Radiography
Plain X-rays show an expansile lytic lesion with variable amounts of internal mineralization. The lesion is usually well-defined with a thin sclerotic rim. Size is characteristically greater than 2cm (distinguishing from osteoid osteoma).
X-ray Findings
- Location: Medullary, eccentric, or cortical
- Pattern: Expansile lytic lesion
- Mineralization: Variable osteoid production
- Rim: Thin sclerotic margin
- Size: Greater than 2cm diameter
- Periosteal reaction: May be present
Spinal Radiographs
- Posterior elements: Pedicle, lamina involvement
- Scoliosis: Painful curve with apical lesion
- Sclerosis: Variable depending on osteoid production
- Soft tissue: May see paraspinal mass
Computed Tomography (CT)
CT is the imaging modality of choice for characterizing osteoblastoma. The classic appearance is mini-brain or lace-like pattern of internal trabeculation with variable mineralization.
CT Imaging Pearl
The "mini-brain appearance" on CT refers to the characteristic lace-like network of trabeculae within the lesion, creating a geographic pattern of mineralization that resembles cerebral gyri. This finding, while not pathognomonic, is highly suggestive of osteoblastoma.
CT Protocol for Osteoblastoma
- Thin slice acquisition (1mm or less)
- Bone and soft tissue windows
- Multiplanar reconstruction (coronal, sagittal)
- 3D reconstruction for surgical planning
- Precise anatomical location and size
- Cortical integrity and expansion
- Internal matrix (lytic, mixed, sclerotic)
- Mini-brain trabecular pattern
- Soft tissue extension
- Neural foraminal involvement
- Spinal canal compromise
- Vertebral body stability
- Relationship to neurovascular structures
Magnetic Resonance Imaging (MRI)
MRI provides superior soft tissue delineation and is essential for spinal lesions to assess neural compression and surgical planning.
MRI Signal Characteristics
| Sequence | Signal Intensity | Clinical Significance |
|---|---|---|
| T1-weighted | Low to intermediate signal | Lesion conspicuity, extent in marrow |
| T2-weighted | High signal (heterogeneous) | Edema, soft tissue extension, ABC component |
| T1 + Gadolinium | Intense enhancement | Hypervascular nature, distinguish from cyst |
| STIR | High signal with edema | Marrow and soft tissue edema extent |
Biopsy
Histological confirmation is mandatory before definitive treatment. Biopsy should be performed by the treating surgeon or musculoskeletal radiologist to ensure proper trajectory that can be excised during definitive surgery.
Biopsy Principles
Essential biopsy considerations:
- CT-guided core needle biopsy preferred (less contamination than open biopsy)
- Biopsy tract must be excisable during definitive procedure
- Multiple cores needed for aggressive variant assessment
- Expert musculoskeletal pathologist review mandatory
- Avoid transarticular or transneural trajectory
Biopsy Technique
- Approach: CT-guided core needle (11-14 gauge)
- Samples: Multiple cores (3-5 specimens)
- Trajectory: Excisable during definitive surgery
- Spinal: Avoid neural structures, consider posterolateral
- Frozen section: Not reliable for diagnosis
Pathology Review
- Expert review: Musculoskeletal pathologist mandatory
- Aggressive features: Assess cellularity, mitoses, atypia
- Differential: Rule out osteosarcoma, ABC, GCT
- Molecular: Consider FOS gene rearrangement if uncertain
- Second opinion: Recommended for aggressive variant
Differential Diagnosis
Key Differentials for Osteoblastoma
| Entity | Age | Location | Size | Pain Pattern | Imaging |
|---|---|---|---|---|---|
| Osteoid osteoma | 10-30y | Long bone cortex | Under 2cm | Night pain, aspirin-responsive | Nidus on CT, intense uptake |
| Osteoblastoma | 10-30y | Spine, long bones | Over 2cm | Progressive, aspirin-resistant | Mini-brain CT, expansile |
| ABC | Under 20y | Metaphysis, spine | Variable | Mild to moderate | Fluid-fluid levels, no osteoid |
| GCT | 20-40y | Epiphysis (closed physis) | Variable | Progressive | Soap bubble, eccentric, lytic |
| Osteosarcoma | 10-25y | Metaphysis | Variable | Rapid progression | Permeative, Codman triangle, sunburst |
Distinguishing Osteoblastoma from Osteoid Osteoma
Key distinguishing features:
Osteoblastoma: Size greater than 2cm, lacks discrete nidus, pain NOT relieved by NSAIDs, often spinal location, expansile growth pattern, intralesional excision treatment.
Osteoid osteoma: Size under 2cm, discrete nidus on CT, night pain dramatically relieved by aspirin/NSAIDs, cortical long bone location, dense sclerosis around nidus, radiofrequency ablation or en bloc excision.
Exam answer: "While both are benign bone-forming tumors with similar histology, osteoblastoma is distinguished by size greater than 2cm, predilection for spine posterior elements, lack of NSAID response, and different treatment approach requiring intralesional or en bloc excision."
Management Algorithm
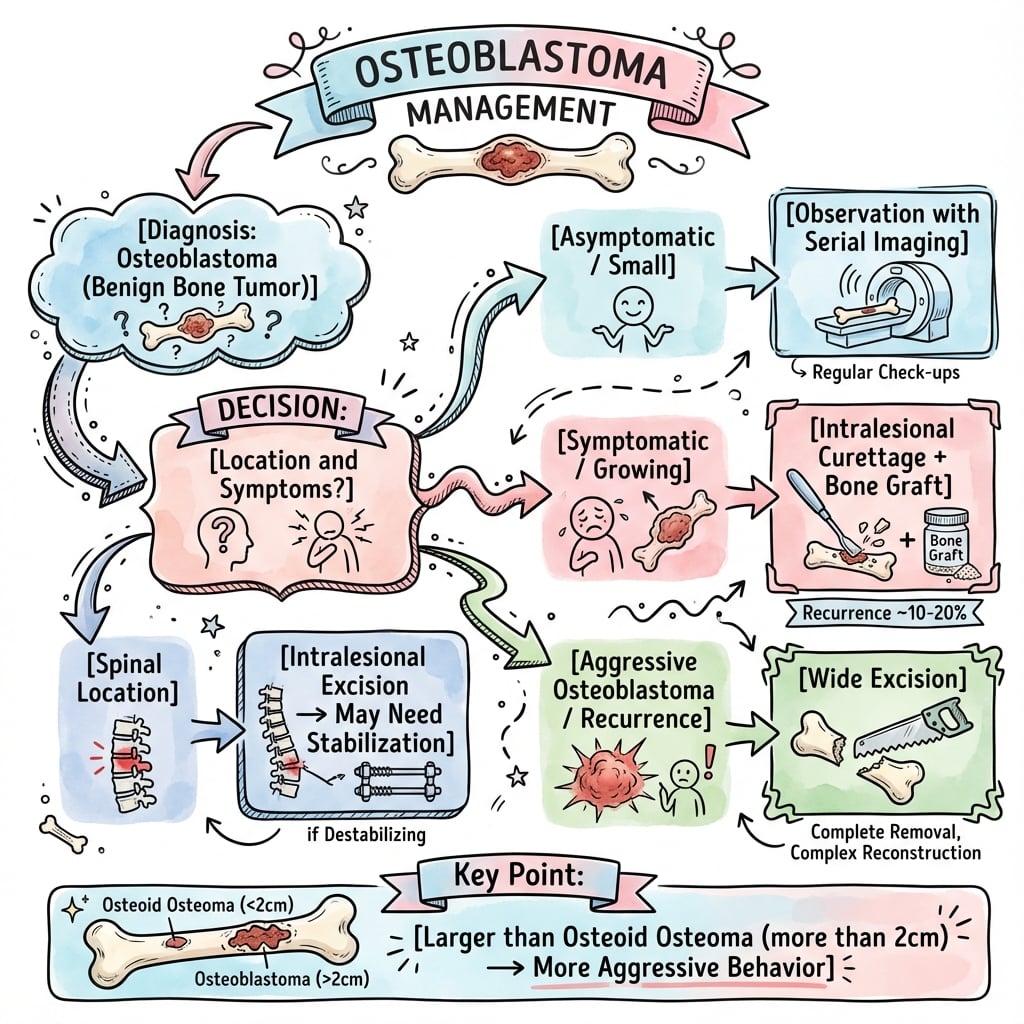
Treatment Algorithm
Stage 2 Active Osteoblastoma
Goal: Complete lesion removal with bone preservation and low recurrence risk.
Surgical Approach
- CT and MRI for precise anatomical mapping
- Tumor board discussion (surgeon, radiologist, pathologist, oncologist)
- Patient counseling regarding recurrence risk (10-20%)
- Planning for reconstruction if needed
- Cortical window for access
- Thorough curettage of entire lesion
- High-speed burr to remove 1-2mm of cavity wall
- Local adjuvant (phenol or argon beam)
- Bone graft (autograft or allograft) or PMMA cement
- PMMA provides thermal adjuvant effect
- Prophylactic fixation if mechanical defect over 50% cortex
- Closure and drain placement
Adjuvant Options
Local adjuvants reduce recurrence by killing microscopic residual tumor:
- Phenol: Applied to cavity wall for 2-3 minutes, then irrigated with alcohol and saline
- PMMA cement: Thermal necrosis from polymerization (reaches 70-80°C)
- Argon beam coagulation: Superficial thermal ablation of cavity
- High-speed burr: Removes 1-2mm margin of cavity wall
Combination of burr plus adjuvant reduces recurrence from 20% to 10%.
Non-Surgical Management
There is no role for chemotherapy or radiation in the primary treatment of osteoblastoma, as it is a benign tumor. These modalities may be considered only in exceptional circumstances.
Non-Surgical Modalities
| Modality | Indication | Efficacy | Role |
|---|---|---|---|
| Observation | Asymptomatic, small, stable lesion | Not curative, risk of growth | Rare, only if surgical risk high |
| Radiation therapy | Unresectable (base of skull, vertebra) | May control growth, risk of malignant transformation | Last resort only |
| Denosumab | Not established for osteoblastoma | Unknown | No role currently |
| Bisphosphonates | Symptomatic pain management | Limited evidence | Adjunct only, not curative |
Complications and Outcomes
Complications
Treatment-Related Complications
| Complication | Incidence | Risk Factors | Management |
|---|---|---|---|
| Local recurrence | 10-20% (intralesional), 25-50% (aggressive) | Incomplete excision, aggressive histology | Revision surgery, consider en bloc |
| Pathological fracture | 5-10% postoperative | Large defect, inadequate fixation | Protected weight-bearing, consider prophylactic fixation |
| Neurological deficit (spine) | 2-5% | Aggressive resection, vascular injury | Immediate decompression, steroids, rehabilitation |
| Wound infection | 2-5% | Prolonged surgery, poor vascularity | Antibiotics, debridement if needed |
| Malignant transformation | Extremely rare (under 1%) | Prior radiation therapy | Wide resection, chemotherapy as for osteosarcoma |
Prognosis and Outcomes
Favorable Prognostic Factors
- Complete excision: Negative margins
- Classic histology: No aggressive features
- Accessible location: Amenable to complete resection
- Small size: Less than 5cm
- Peripheral skeleton: Hand, foot (easier excision)
Poor Prognostic Factors
- Aggressive histology: Epithelioid features, increased mitoses
- Incomplete excision: Residual tumor
- Spinal location: Difficult complete excision
- Large size: Greater than 10cm
- Recurrent disease: Higher re-recurrence rate
Surveillance Protocol
Post-treatment surveillance:
- Clinical examination every 3 months for first year, then 6 monthly for 2 years
- Plain X-rays at each visit
- MRI at 3 months, 1 year, 2 years (more sensitive for recurrence)
- CT if X-ray suspicious for recurrence
- Recurrence typically within first 2 years (90% of recurrences)
- After 5 years disease-free, discharge to primary care
Evidence Base and Key Studies
Osteoblastoma: clinicopathologic study of 306 cases
- 306 osteoblastomas analysed (Mayo Clinic plus consultation files); mean age 20.4 years, male:female 2:1
- Vertebral column including sacrum was the single most frequent site (32% of cases)
- Recurrence 16% (Mayo cases) and 21% (consultation cases); central neuraxis tumours carried greater morbidity and mortality
- Large epithelioid osteoblasts seen in 24% and predominant in 10%; 12% had radiographic features suggesting malignancy
- Histopathology alone did not reliably predict aggressive behaviour; osteosarcoma is the key differential
Borderline osteoblastic tumors: problems in the differential diagnosis of aggressive osteoblastoma and low-grade osteosarcoma
- Reviewed 102 benign osteoblastic tumours; classified into osteoid osteoma, osteoblastoma and aggressive osteoblastoma (15 cases)
- Defined histological criteria for aggressive osteoblastoma and the term itself, distinct from low-grade osteosarcoma
- Proposed four categories of confusing osteoblastic lesions, with low-grade osteosarcoma mimicking osteoblastoma the commonest source of diagnostic error
- Locally aggressive osteoblastomas recur but do not metastasise; a minority truly transform to osteosarcoma in recurrences
- Established that pseudosarcomatous histology can coexist with a benign clinical course
Recurrent rearrangements of FOS and FOSB define osteoblastoma
- Whole-genome DNA and RNA sequencing identified FOS rearrangement in 5 tumours and FOSB rearrangement in 1
- Extended to a cohort of 55 cases using FISH and immunohistochemistry showing near-ubiquitous FOS or FOSB mutation
- Established osteoblastoma and osteoid osteoma as a single molecularly defined family of FOS/FOSB-driven bone tumours
- FOS immunohistochemistry provides a practical diagnostic adjunct to distinguish from osteosarcoma, which lacks these rearrangements
- Resolved the long-standing paradox that v-fos causes murine osteosarcoma yet FOS point mutations are absent in human bone tumours
An update of molecular pathology of bone tumors: lessons from next generation sequencing
- Catalogues mutually exclusive defining alterations across benign and malignant bone tumours
- Osteoblastoma and osteoid osteoma are defined by FOS/FOSB rearrangements
- Contrasts with ABC (USP6), chondroblastoma (H3F3B K36M) and giant cell tumour (H3F3A G34W)
- High-grade osteosarcoma notably lacks a single recurrent defining alteration, aiding the osteoblastoma differential
- Many markers translated to clinical FISH or immunohistochemistry assays
Osteoblastoma: clinical and radiologic findings in 98 new cases
- 98 new osteoblastoma patients with radiograph, CT and MRI correlation
- Typically a lytic medullary lesion with matrix ossification and mild surrounding sclerosis
- CT best demonstrated tumour origin, extent, matrix mineralisation and the thin bony shell
- MRI added separation of tumour tissue from surrounding marrow and soft-tissue oedema
- In hands, feet and skull the appearance overlapped with ABC, giant cell tumour and button sequestrum
Diagnostic and management options of osteoblastoma in the spine
- 13 surgically treated spinal osteoblastomas, mean follow-up 43.8 months, treated without postoperative radiotherapy
- VAS pain fell from 6.2 to 0.5 and Oswestry Disability Index from 51.1 to 22.6 (both p less than 0.001)
- Every patient with a preoperative deficit (ASIA C/D) recovered to ASIA E by final follow-up
- Only 1 of 13 recurred, managed by en bloc marginal resection with postoperative radiotherapy
- Accurate intraoperative localisation and complete resection were identified as the keys to preventing recurrence
Exam Viva Scenarios
Use these scenarios to practise clinical reasoning and management decisions
Scenario 1: Initial Diagnosis and Classification
"A 16-year-old male presents with 4 months of progressive left thigh pain, worse at night but not relieved by ibuprofen. X-ray shows a 3cm expansile lytic lesion in the proximal femur metaphysis with internal mineralization and a thin sclerotic rim. How would you assess and manage this patient?"
Scenario 2: Aggressive Variant Histology
"The biopsy of the proximal femur lesion shows osteoid production by epithelioid osteoblasts with increased mitotic activity (3-4 per HPF) and sheet-like growth pattern. The radiologist is concerned about osteosarcoma. How do you proceed?"
Scenario 3: Spinal Osteoblastoma with Neurological Deficit
"A 14-year-old female presents with 3 months of back pain and 2 weeks of progressive right leg weakness (grade 3/5 power). MRI shows a 4cm expansile lesion arising from the L3 left pedicle and lamina with 50% spinal canal compromise and cord compression. How would you manage this patient?"
MCQ Practice Points
Size Criterion Question
Q: What is the size threshold that distinguishes osteoblastoma from osteoid osteoma? A: Greater than 2cm. Osteoblastoma is by definition larger than 2cm, while osteoid osteoma is smaller than 2cm. This size criterion, combined with clinical features (NSAID response) and imaging (nidus presence), helps distinguish these histologically similar tumors.
Anatomical Predilection Question
Q: What percentage of osteoblastomas occur in the spine, and which anatomical structures are most commonly involved? A: About 30-40% occur in the spine (32% in the 306-case Lucas series), most commonly affecting the posterior elements (pedicle, lamina, and transverse process). This predilection for spine distinguishes osteoblastoma from osteoid osteoma, which favors long bone cortical locations.
Histology Question
Q: What histological features distinguish aggressive osteoblastoma from conventional osteosarcoma? A: Aggressive osteoblastoma has: (1) circumscribed margin (not permeative), (2) uniform osteoblasts (not pleomorphic), (3) sheet-like growth but organized, (4) increased mitoses (2-5 per HPF) but not as high as osteosarcoma. Despite aggressive histology, it does NOT metastasize and is benign.
Treatment Question
Q: What is the recurrence rate of osteoblastoma after intralesional curettage, and how can it be reduced? A: 10-20% recurrence after intralesional curettage alone. Recurrence can be reduced to approximately 10% by using adjuvants: high-speed burr (removes 1-2mm cavity margin), local phenol application, and PMMA cement (thermal necrosis). Aggressive variant has 25-50% recurrence with curettage, requiring en bloc resection.
Imaging Question
Q: What is the characteristic CT appearance of osteoblastoma? A: Mini-brain or lace-like pattern of internal trabeculation within an expansile lytic lesion. This refers to the geographic network of osteoid and woven bone creating a pattern resembling cerebral gyri. Also shows variable mineralization and thin sclerotic rim.
Management Question
Q: Is there a role for chemotherapy or radiation in the primary treatment of aggressive osteoblastoma? A: No. Osteoblastoma is a benign tumor that does NOT metastasize, even the aggressive variant. Treatment is surgical (intralesional with adjuvants or en bloc resection). Chemotherapy and radiation are NOT indicated except in extremely rare unresectable cases (base of skull, cervical vertebra) where radiation may be considered as last resort.
Areas of Controversy and Uncertainty
Osteoblastoma generates several genuine debates that examiners use to probe depth of understanding.
Osteoblastoma vs low-grade osteosarcoma
The hardest distinction is not from osteoid osteoma but from osteoblastoma-like (low-grade) osteosarcoma, which can show near-identical biopsy morphology. The safeguard is concordance of clinical, radiological and pathological data plus, increasingly, FOS/FOSB testing (positive in osteoblastoma, negative in osteosarcoma). Where there is discordance, treat as malignant until proven otherwise.
Is 'aggressive osteoblastoma' a real entity?
Lucas et al. found histology alone did not reliably predict recurrence, questioning whether epithelioid or aggressive osteoblastoma is a distinct biological category or simply the upper end of a spectrum. Most now treat radiological aggressiveness and incomplete excision, rather than the histological label, as the operative drivers of management.
Intralesional vs en bloc excision
For accessible expendable bone, en bloc resection minimises recurrence but is rarely justified for a benign lesion. Intralesional curettage with adjuvants is standard for most lesions; en bloc is reserved for aggressive radiology, recurrence, or where complete curettage is impossible. There is no randomised evidence comparing the two.
Role of adjuvants and ablation
Phenol, PMMA and burring are widely used but supported only by retrospective data; their independent contribution beyond complete curettage is unproven. Percutaneous thermal ablation (radiofrequency, cryoablation) is established for osteoid osteoma but its role in larger osteoblastoma, especially near neural structures, remains investigational.
Guidelines, Registries & Global Practice
Global epidemiology: Osteoblastoma is rare, accounting for about 1% of all primary bone tumours and roughly 3% of benign bone tumours, with a consistent worldwide picture of peak incidence in the second and third decades and a male predominance of about 2:1. The spinal predilection (30-40%, 32% in the largest series) is reported across North American, European and Asian cohorts, indicating no major geographic variation in biology.
Side-by-side guidance: There is no osteoblastoma-specific guideline; practice is governed by general bone-tumour pathways.
How major frameworks frame benign aggressive bone tumours
| Body | Position relevant to osteoblastoma |
|---|---|
| WHO Classification of Bone Tumours (5th ed) | Lists osteoblastoma as benign; recognises FOS/FOSB rearrangement as the defining molecular feature shared with osteoid osteoma |
| ESMO/EURACAN/SSG sarcoma guidance (Europe) | Any bone lesion with an uncertain or aggressive appearance should be referred to a bone-sarcoma centre for biopsy and MDT diagnosis before definitive surgery |
| NICE / BOA-BOAST (UK) | Suspected primary bone tumours referred to a specialist supraregional bone-tumour service; biopsy performed at or directed by the treating centre |
| NCCN bone cancer (US) | Emphasises image-guided biopsy with an excisable tract and multidisciplinary review before resection of any indeterminate bone lesion |
Registries: Osteoblastoma is benign and is not tracked by the arthroplasty registries (NJR, AJRR, AOANJRR, SHAR); incidence and outcome data come instead from national bone-tumour and pathology reference centres (for example the Mayo, Rizzoli and Royal National Orthopaedic Hospital series).
High- vs limited-resource practice: In high-resource settings, thin-slice CT, MRI, image-guided biopsy, FOS/FOSB ancillary testing and specialist musculoskeletal pathology are standard, allowing confident separation from osteosarcoma before surgery. In limited-resource settings, diagnosis may rest on plain radiographs and conventional histology, raising the risk of either under-treating a low-grade osteosarcoma or over-treating a benign lesion; referral of indeterminate bone-forming lesions to a tumour centre remains the universal safeguard.
OSTEOBLASTOMA
Clinical summary
Key Features
- •Benign bone-forming tumor, 1% of primary bone tumors, 3% of benign
- •Peak age 10-30 years, male greater than female 2:1
- •Greater than 2cm size (distinguishes from osteoid osteoma)
- •40% spine (posterior elements: pedicle, lamina), 30% long bones
- •Pain NOT relieved by NSAIDs (unlike osteoid osteoma)
Imaging
- •X-ray: Expansile lytic lesion, variable mineralization, thin sclerotic rim
- •CT: Mini-brain (lace-like) trabecular pattern - highly characteristic
- •MRI: Low T1, high T2, intense enhancement, marrow edema
- •No discrete nidus (unlike osteoid osteoma)
- •Biopsy: CT-guided core needle, excisable trajectory
Histology
- •Osteoid and woven bone production by active osteoblasts
- •Vascular fibrous stroma, osteoclast-like giant cells
- •Aggressive variant: epithelioid osteoblasts, sheet-like growth, increased mitoses
- •Distinguish from osteosarcoma: circumscribed margin, uniform cells, no permeation
- •Expert musculoskeletal pathology review mandatory
Treatment Algorithm
- •Stage 2 (Active): Intralesional curettage + burr + adjuvant (phenol or PMMA)
- •Stage 3 (Aggressive): En bloc wide resection with 1-2cm margin
- •Spinal: Decompression + tumor excision + instrumented fusion if destabilizing
- •NO chemotherapy or radiation (benign tumor)
- •Recurrence 10-20% (intralesional), 25-50% (aggressive with curettage)
Differential Diagnosis
- •Osteoid osteoma: Under 2cm, nidus, NSAID-responsive, cortical long bone
- •ABC: Fluid-fluid levels, no osteoid production, younger age
- •GCT: Epiphyseal, older age (20-40y), soap bubble appearance
- •Osteosarcoma: Permeative, Codman triangle, atypical cells, rapid growth
- •Chondroblastoma: Epiphyseal, chondroid matrix, younger age