X-Linked Hypophosphataemia (XLH) and Tumour-Induced Osteomalacia (TIO)
- FGF23 is a phosphaturic hormone made by osteocytes; its EXCESS causes RENAL PHOSPHATE WASTING (it downregulates the proximal-tubule sodium-phosphate cotransporters) AND suppresses 1-alpha-hydroxylase, giving an inappropriately LOW (or normal) 1,25-dihydroxyvitamin D - the combination produces HYPOPHOSPHATAEMIA and impaired mineralization (RICKETS in children, OSTEOMALACIA in adults).
- X-LINKED HYPOPHOSPHATAEMIA (XLH) is the commonest heritable form: an X-linked dominant PHEX gene mutation leads to excess FGF23. Children present with RICKETS, lower-limb BOWING (genu varum/valgum), short stature, bone pain, dental abscesses and enthesopathy; adults have osteomalacia, pseudofractures, enthesopathy and early osteoarthritis.
- TUMOUR-INDUCED (ONCOGENIC) OSTEOMALACIA (TIO) is an ACQUIRED paraneoplastic syndrome in which a usually small, benign PHOSPHATURIC MESENCHYMAL TUMOUR secretes FGF23, causing adult-onset osteomalacia with bone pain, muscle weakness and fractures; the great challenge is LOCALISING the small tumour (functional imaging such as 68Ga-DOTATATE PET/CT, octreotide scintigraphy, and selective venous sampling).
- The BIOCHEMISTRY is distinctive: LOW serum PHOSPHATE with renal phosphate wasting (low TmP/GFR, high fractional phosphate excretion), inappropriately LOW or normal 1,25-vitamin D, NORMAL calcium, NORMAL or HIGH alkaline phosphatase (osteomalacia), normal/mildly raised PTH, and elevated FGF23 - importantly the ALP is NOT low (contrast with hypophosphatasia, where ALP is LOW).
- CONVENTIONAL treatment is oral PHOSPHATE supplements plus ACTIVE vitamin D (calcitriol/alfacalcidol), but this is burdensome and risks GI upset, secondary/tertiary hyperparathyroidism and nephrocalcinosis; the targeted therapy BUROSUMAB, a monoclonal antibody against FGF23, addresses the underlying mechanism and is first-line for XLH (children and adults) and for inoperable/metastatic TIO.
- For TIO, complete SURGICAL RESECTION of the FGF23-secreting tumour is the DEFINITIVE, curative treatment (FGF23, phosphate and symptoms normalise); when the tumour cannot be found or resected, burosumab or conventional phosphate/active-vitamin-D therapy is used. Orthopaedic care addresses pseudofractures, deformity (osteotomy timed around medical optimisation) and enthesopathy.
- “XLH (PHEX, inherited) and TIO (FGF23 tumour, acquired) both = EXCESS FGF23 -> renal phosphate wasting + low/normal 1,25-vitD -> rickets/osteomalacia.
- “Biochemistry: LOW phosphate + phosphate wasting + low/normal 1,25-vitD + NORMAL calcium + NORMAL/high ALP + high FGF23 (ALP NOT low - unlike hypophosphatasia).
- “TIO: localise the tumour (68Ga-DOTATATE PET) -> resection is curative. Burosumab (anti-FGF23) is first-line for XLH and inoperable TIO.
Low phosphate + renal phosphate wasting + low/normal 1,25-vitD + NORMAL/high ALP + high FGF23. Treat with phosphate/active vitamin D, burosumab, and (TIO) tumour resection.
Hypophosphatasia also has a 'hypophosphat-' name but a LOW ALP and is treated with asfotase alfa (and bisphosphonates are contraindicated). (See our Hypophosphatasia topic.)
Mechanism: Too Much FGF23
FGF23, secreted by osteocytes, normally keeps phosphate in check; in excess it does two things that cause disease: it downregulates the sodium-phosphate cotransporters in the proximal renal tubule, causing renal phosphate wasting and a low serum phosphate, and it suppresses 1-alpha-hydroxylase, so 1,25- dihydroxyvitamin D is inappropriately low or normal (it should rise in response to low phosphate). The result is chronic hypophosphataemia and a failure of bone mineralization - rickets in the growing child and osteomalacia in the adult. In XLH the FGF23 excess is driven by an inherited PHEX mutation; in TIO it is driven by an FGF23-secreting tumour.
XLH vs TIO & Workup
- XLH: suspect in a child with rickets, lower-limb bowing, short stature, dental abscesses or a family history (X-linked dominant PHEX); confirm with the phosphate-wasting biochemistry and genetics. Adults have osteomalacia, pseudofractures and enthesopathy.
- TIO: suspect in an adult with new, unexplained osteomalacia, severe hypophosphataemia, bone pain, weakness and fractures, and no family history; the tumour is usually a small, benign phosphaturic mesenchymal tumour in bone or soft tissue (often extremities/head and neck) that is hard to find - functional imaging (68Ga-DOTATATE PET/CT, octreotide scintigraphy) and selective venous sampling for FGF23 help localise it.
- Biochemistry (both): low phosphate with renal phosphate wasting (low TmP/GFR, high fractional excretion), inappropriately low/normal 1,25-vitamin D, normal calcium, normal/high ALP, normal/mildly raised PTH, and elevated FGF23 - the normal/high ALP distinguishes these from hypophosphatasia.
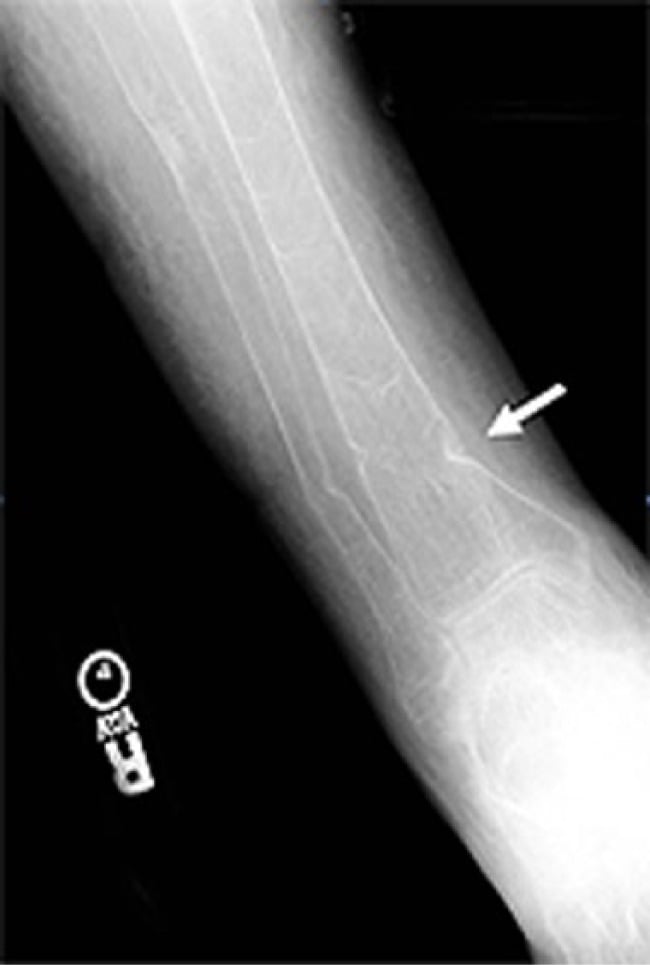
- Radiographs show rickets (children: physeal widening/cupping/bowing) or osteomalacia with Looser-zone pseudofractures (adults).
The Hypophosphataemic-Rickets Differential
- FGF23-mediated (XLH, TIO, and also fibrous dysplasia / McCune-Albright and the autosomal hypophosphataemic rickets): renal phosphate wasting, inappropriately low/normal 1,25-vitamin D, high FGF23, normal calcium.
- Nutritional / vitamin-D-deficient rickets/osteomalacia: a different mechanism - low 25-vitamin D, low/low-normal calcium, and secondary hyperparathyroidism (high PTH) that itself drives the phosphaturia; responds to vitamin D and calcium.
- Hereditary hypophosphataemic rickets with hypercalciuria (HHRH, SLC34A3): renal phosphate wasting that is FGF23-independent, with an appropriately HIGH 1,25-vitamin D and hypercalciuria - treated with phosphate alone (active vitamin D is not needed and would worsen the hypercalciuria).
- Fanconi syndrome / proximal tubulopathy: phosphate wasting plus glycosuria, aminoaciduria and bicarbonate loss (a generalised proximal-tubule defect).
- Hypophosphatasia: the low-ALP mimic (treated with asfotase alfa).
The discriminators: check PTH, calcium, 25- and 1,25-vitamin D, FGF23 and a urine screen. High FGF23 with low/normal 1,25-vitamin D and normal calcium points to XLH/TIO; high PTH with low calcium and low 25-vitamin D points to nutritional disease; a high 1,25-vitamin D with hypercalciuria points to HHRH. (General rickets/osteomalacia and vitamin-D deficiency are covered in our Rickets, Osteomalacia and Vitamin D Deficiency topics.)
For a low phosphate, let PTH/calcium/1,25-vitamin D sort it: FGF23 high + low/normal 1,25-vitamin D + normal calcium = XLH/TIO; high PTH + low calcium + low 25-vitamin D = nutritional; high 1,25-vitamin D + hypercalciuria = HHRH (phosphate alone); glycosuria/aminoaciduria = Fanconi; low ALP = hypophosphatasia.
Management
- Conventional medical therapy: oral phosphate supplements (divided doses) plus active vitamin D (calcitriol/alfacalcidol) - effective but burdensome, with risks of GI upset, secondary/tertiary hyperparathyroidism and nephrocalcinosis; monitor calcium, PTH and renal tract.
- Burosumab (anti-FGF23 monoclonal antibody): targets the underlying excess FGF23 and is first-line for XLH in children and adults, healing rickets/osteomalacia and improving symptoms; also used for inoperable/metastatic TIO.
- TIO - surgery: complete resection of the FGF23-secreting tumour is definitive and curative - FGF23, phosphate and symptoms normalise; when the tumour cannot be localised/resected, use burosumab or conventional therapy.
- Orthopaedic care: manage pseudofractures, correct deformity (e.g. tibial/femoral osteotomy, timed around medical optimisation to aid union), and address enthesopathy and secondary osteoarthritis; optimise phosphate/vitamin D peri-operatively to support healing.
In an adult with new osteomalacia and severe hypophosphataemia, actively pursue a hidden FGF23-secreting tumour, because resection is curative and the disease otherwise persists - use functional imaging and venous sampling rather than giving up after negative cross-sectional scans. And be precise with the biochemistry: a LOW phosphate with a normal or high alkaline phosphatase points to an FGF23 disorder, whereas a LOW alkaline phosphatase points to hypophosphatasia - two very different diseases with opposite treatments.
The Orthopaedic Burden & Deformity Surgery
- Lower-limb deformity (genu varum/valgum, tibial/femoral bowing and torsion): in the growing child, guided growth (hemiepiphysiodesis) can correct angular deformity while growth remains; in the adult or severe/multi-apical deformity, corrective osteotomy.
- The cardinal principle: correct deformity only after medical optimisation (phosphate/active vitamin D or burosumab), because operating on poorly-mineralised, biochemically-uncontrolled bone risks delayed union/nonunion, hardware failure and rapid deformity recurrence.
- Pseudofractures (Looser zones) largely heal with medical control; an impending or displaced one may need fixation.
- Enthesopathy - progressive ossification/calcification of tendon and ligament insertions - is characteristic of adult XLH and causes pain and stiffness; in the spine it (with facet hypertrophy and ligament ossification) can cause spinal canal stenosis and cord/cauda compression, a recognised XLH complication to watch for.
- Plus early osteoarthritis, dental abscesses, and - from conventional therapy - iatrogenic nephrocalcinosis and hyperparathyroidism.
(Guided-growth and osteotomy technique are covered in our Guided Growth and Osteotomy topics, and general spinal stenosis in our Lumbar Spinal Stenosis topic; the point here is the XLH-specific burden and the timing rule.)
Correct deformity only after the biochemistry is optimised (phosphate/active vitamin D or burosumab) - operating on uncontrolled bone risks nonunion and recurrence. Use guided growth in the growing child, osteotomy in the adult; watch for enthesopathy (and XLH spinal stenosis) and conventional-therapy nephrocalcinosis/hyperparathyroidism.
Mnemonics & Memory Aids
FGF23
Hook:FGF23 excess wastes phosphate and lowers active vitamin D.
ALP TELLS
Hook:Let the ALP tell you: normal/high = FGF23 (XLH/TIO); low = hypophosphatasia.
Clinical Decision Scenarios
Practise clinical reasoning and management decisions out loud
“An adult presents with osteomalacia, bone pain and severe hypophosphataemia with renal phosphate wasting. What is the likely mechanism, what would you look for, and how is it treated?”
“How does X-linked hypophosphataemia present and how is it managed, and how does the biochemistry differ from hypophosphatasia?”
Mechanism
- Excess FGF23 -> renal phosphate wasting + suppressed 1,25-vitamin D
- -> hypophosphataemia -> rickets (children) / osteomalacia (adults)
- XLH = PHEX (inherited); TIO = FGF23-secreting tumour (acquired)
Biochemistry
- LOW phosphate + renal phosphate wasting (low TmP/GFR)
- Inappropriately low/normal 1,25-vitD; normal calcium; NORMAL/high ALP; high FGF23
- ALP NOT low (contrast with hypophosphatasia)
Diagnosis
- XLH: childhood rickets/bowing + family history (PHEX) + genetics
- TIO: adult osteomalacia, no family history; localise tumour (68Ga-DOTATATE PET, venous sampling)
- Radiographs: rickets (child) / Looser-zone pseudofractures (adult)
Management
- Conventional: oral phosphate + active vitamin D (monitor PTH/nephrocalcinosis)
- Burosumab (anti-FGF23): first-line for XLH; inoperable TIO
- TIO: tumour resection is curative; osteotomy for deformity (optimise phosphate first)
Evidence & Key Studies
X-linked hypophosphatemia and tumour-induced osteomalacia in the era of burosumab: a narrative review
- XLH and TIO are both caused by persistent excess FGF23 leading to renal phosphate wasting and reduced phosphate availability.
- Traditional oral phosphate and active vitamin D have limitations; burosumab directly targets FGF23 and is recommended first-line for XLH (1-17 years and adults).
- For TIO, tumour resection is the definitive treatment (success depends on localisation and surgical expertise), with burosumab or conventional therapy when surgery is not feasible.
Burosumab for FGF23-associated hypophosphatemia in fibrous dysplasia/McCune-Albright syndrome
- FGF23 overproduction can cause hypophosphataemia beyond XLH/TIO - here in fibrous dysplasia/McCune-Albright syndrome.
- Burosumab (anti-FGF23) normalised calcium-phosphate metabolism and markedly reduced alkaline phosphatase, with no further fractures over 24 months.
- Demonstrates the central role of FGF23 and the efficacy of targeting it, while noting more data are needed in non-XLH settings.
The shared FGF23 mechanism of XLH and TIO, the first-line role of burosumab for XLH and the curative role of tumour resection for TIO come from the cited Brandi review, and the central role of FGF23 and the response to burosumab from the cited Stelmachowska-Banas case. The phosphate-wasting biochemistry, the contrast with hypophosphatasia's low ALP, and the rickets/osteomalacia radiology are standard, well-established teaching. (See also our Hypophosphatasia, Rickets/Osteomalacia and Calcium/Metabolic Bone topics.)