The 'Stippled Epiphyses' Dysplasias
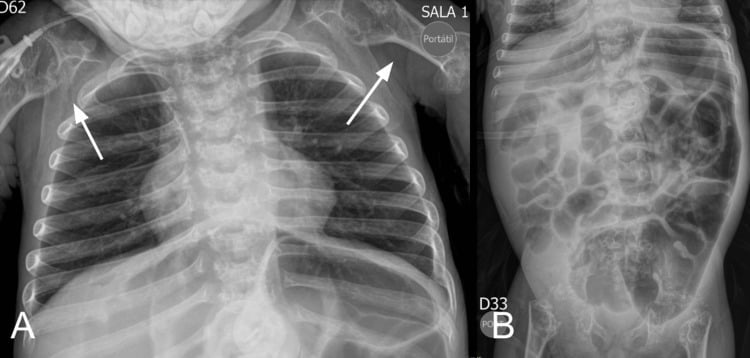
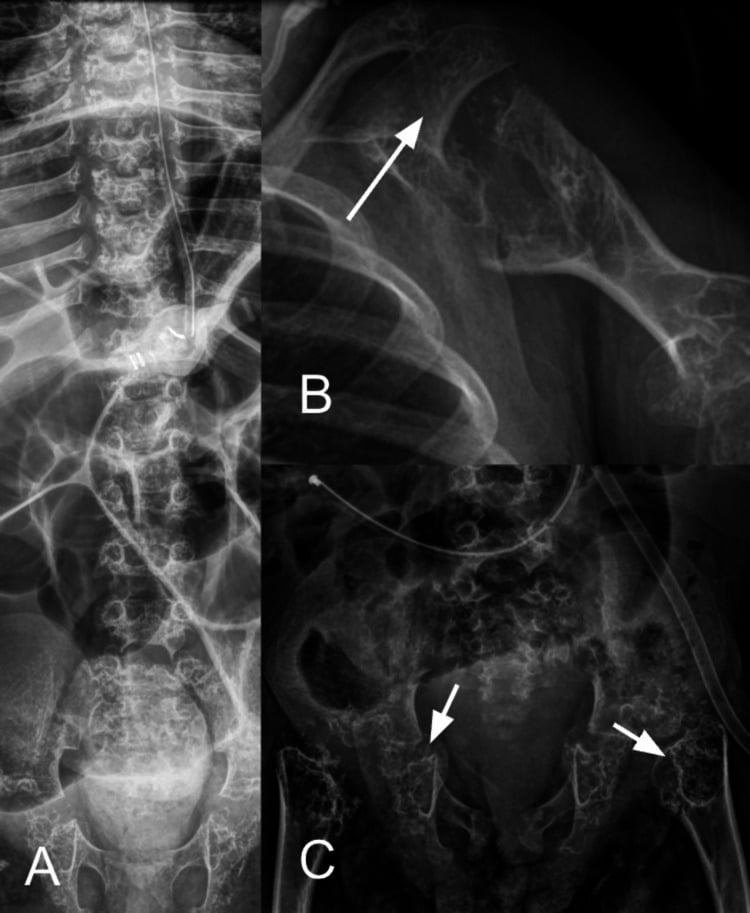
- CHONDRODYSPLASIA PUNCTATA (CDP) is a heterogeneous GROUP of skeletal dysplasias unified by a single radiographic HALLMARK in infancy: STIPPLED (punctate) epiphyseal and peri-articular CALCIFICATION (the calcific stippling characteristically RESOLVES as the child grows); chondrodysplasia punctata is recognised among the skeletal dysplasias diagnosed in the fetal/neonatal period.
- The RHIZOMELIC form is the most severe, but it is a SPECTRUM rather than one disease: an AUTOSOMAL-RECESSIVE peroxisomal disorder of PLASMALOGEN synthesis (five types - PEX7, GNPAT, AGPS, FAR1 and the long isoform of PEX5). CLASSIC disease, with negligible plasmalogens, gives symmetric rhizomelic shortening, profound developmental delay, congenital cataracts, contractures and often early death; NONCLASSIC patients with HIGHER residual plasmalogen levels have better growth and cognition and survive longer - so the plasmalogen level is prognostic, not merely diagnostic, and counselling should wait for it.
- CONRADI-HUNERMANN syndrome (CDPX2) is X-LINKED DOMINANT (EBP gene), affects mostly FEMALES (often lethal in males), and is milder/survivable: it causes ASYMMETRIC limb shortening (and hence limb-length DISCREPANCY), SCOLIOSIS, ICHTHYOSIS/skin changes following Blaschko's lines, and CATARACTS - the asymmetry is a key clue.
- There is also an X-LINKED RECESSIVE (brachytelephalangic) form (ARSL/arylsulfatase defects) with nasal and distal-phalangeal hypoplasia, and important SECONDARY/TERATOGENIC causes that phenocopy CDP - notably WARFARIN EMBRYOPATHY (maternal warfarin in pregnancy) and maternal AUTOIMMUNE disease (e.g. systemic lupus erythematosus) - so a maternal drug/autoimmune history matters.
- The ORTHOPAEDIC sequelae depend on the form and include RHIZOMELIC/asymmetric limb SHORTENING and limb-length DISCREPANCY, SCOLIOSIS (especially Conradi-Hunermann), joint CONTRACTURES and (in milder survivors) deformity; ophthalmology (cataracts) and the systemic/metabolic aspects are co-managed.
- MANAGEMENT is largely SUPPORTIVE and form-dependent within a multidisciplinary team: genetic/metabolic diagnosis (distinguishing peroxisomal rhizomelic CDP from EBP/ARSL forms and from teratogenic causes), ORTHOPAEDIC management of limb-length discrepancy (epiphysiodesis/lengthening), SCOLIOSIS and contractures in survivable forms, cataract surgery, and family counselling - with the severe rhizomelic form being largely palliative.
- “Chondrodysplasia punctata = heterogeneous group; HALLMARK = STIPPLED (punctate) epiphyseal calcification in infancy (resolves with age).
- “RHIZOMELIC CDP = AUTOSOMAL-RECESSIVE PEROXISOMAL plasmalogen defect (RCDP1-5: PEX7, GNPAT, AGPS, FAR1, PEX5L) - a SPECTRUM graded by residual plasmalogen: classic (negligible) severe with early death, NONCLASSIC (higher) milder with better growth and cognition. CONRADI-HUNERMANN (CDPX2) = X-linked DOMINANT (EBP), females, ASYMMETRIC limb shortening + ichthyosis + cataracts.
- “Also X-linked recessive (brachytelephalangic) + TERATOGENIC phenocopies (WARFARIN embryopathy, maternal SLE/autoimmune). Orthopaedic: limb shortening/discrepancy, scoliosis, contractures - supportive + deformity correction.
Infant radiograph: stippled (punctate) epiphyses. Rhizomelic (AR peroxisomal) = severe/symmetric; Conradi-Hunermann (X-linked dominant, EBP) = milder/asymmetric + ichthyosis + cataracts.
Teratogenic phenocopies: maternal warfarin (warfarin embryopathy) and maternal autoimmune disease (SLE). Take a maternal drug/autoimmune history.
The Forms & Their Features
Chondrodysplasia punctata is a heterogeneous group unified by stippled (punctate) epiphyseal calcification on infant radiographs (which resolves with growth). The rhizomelic form is a severe autosomal-recessive peroxisomal plasmalogen disorder spanning RCDP1 to RCDP5, in which classic disease gives symmetric rhizomelia, cataracts, contractures and often early death while nonclassic patients with higher residual plasmalogens do considerably better. Conradi-Hunermann (CDPX2) is X-linked dominant (EBP), mostly in females, milder, with asymmetric limb shortening (limb-length discrepancy), scoliosis, ichthyosis (along Blaschko's lines) and cataracts. An X-linked recessive (brachytelephalangic) form and teratogenic phenocopies - warfarin embryopathy and maternal autoimmune disease (SLE) - complete the group, so a maternal drug/autoimmune history is important.
- Genetics
- Autosomal recessive peroxisomal: PEX7, GNPAT, AGPS, FAR1, PEX5L
- Key features
- A spectrum graded by residual plasmalogen - classic: symmetric rhizomelia, delay, cataracts, early death; nonclassic: milder, better growth and cognition
- Genetics
- X-linked dominant (EBP); mostly females
- Key features
- Asymmetric limb shortening (discrepancy), scoliosis, ichthyosis, cataracts; milder
- Genetics
- ARSL/arylsulfatase
- Key features
- Brachytelephalangic - nasal + distal-phalangeal hypoplasia
- Genetics
- Warfarin embryopathy; maternal autoimmune (SLE)
- Key features
- Phenocopy of CDP - take maternal history
Warfarin Embryopathy and the Fuller Differential
- Warfarin embryopathy - the mechanism. Warfarin crosses the placenta and inhibits vitamin-K epoxide reductase, impairing γ-carboxylation of the vitamin-K-dependent bone protein matrix Gla protein (a normal inhibitor of cartilage calcification) - so cartilage calcifies abnormally, giving stippled epiphyses and nasal hypoplasia (fetal warfarin syndrome). The critical teratogenic window is the first trimester (about 6-9 weeks), which is why heparin/LMWH (which do not cross the placenta) are substituted in pregnancy.
- Other secondary causes. Maternal autoimmune disease (SLE), maternal vitamin-K deficiency and some anticonvulsants (phenytoin) can also produce a stippled-epiphyses phenocopy.
- The fuller differential of stippled epiphyses. Beyond CDP, consider Zellweger spectrum (another peroxisomal disorder), congenital hypothyroidism (epiphyseal dysgenesis), trisomy 18/21, GM1 gangliosidosis, congenital infection, and the cholesterol-pathway disorders CHILD syndrome (NSDHL) and Smith-Lemli-Opitz (DHCR7).
Q: What is the mechanism of warfarin embryopathy, and what is the fuller differential of stippled epiphyses?
A: Warfarin inhibits vitamin-K epoxide reductase → impaired γ-carboxylation of matrix Gla protein (a normal inhibitor of cartilage calcification) → abnormal cartilage calcification = stippled epiphyses + nasal hypoplasia (fetal warfarin syndrome); the critical window is the first trimester (~6-9 weeks) → substitute heparin/LMWH. Other causes: maternal SLE, vitamin-K deficiency, anticonvulsants. Fuller differential: Zellweger, congenital hypothyroidism (epiphyseal dysgenesis), trisomy 18/21, GM1 gangliosidosis, CHILD (NSDHL), Smith-Lemli-Opitz (DHCR7).
Rhizomelic CDP Is a Spectrum, Not a Single Lethal Disease
Calling the rhizomelic form uniformly severe and early-lethal is the traditional teaching and is wrong at the milder end. Severe (classic) RCDP has negligible plasmalogen levels with congenital cataracts, skeletal dysplasia, growth failure and neurodevelopmental deficit. Individuals with milder or nonclassic RCDP have higher residual plasmalogen levels, better growth and better cognitive outcomes, and survive well beyond infancy. The plasmalogen deficiency is itself described as the main pathogenic factor determining severity - so the red-cell plasmalogen level is not merely a diagnostic test but a prognostic one, and a family should not be counselled from the classic phenotype before the biochemistry is known.
Rhizomelic CDP is subdivided by the step that fails in plasmalogen synthesis: RCDP1 - PEX7 (the PTS2 receptor), RCDP2 - GNPAT, RCDP3 - AGPS, RCDP4 - FAR1, and RCDP5 - loss of the long isoform of PEX5 (PEX5L), reported in four patients from two families. RCDP5 matters conceptually: other PEX5 mutations cause Zellweger spectrum disease, but losing only PEX5L knocks out import of PTS2-tagged proteins alone - the same functional lesion as PEX7 - so it produces RCDP instead. Because RCDP1 and RCDP5 overlap biochemically, a selective PTS2-import defect warrants sequencing both PEX7 and PEX5 exon 9.
In the severe phenotype, MRI shows supratentorial white-matter abnormality with a parieto-occipital predominance, and cerebellar atrophy appears in older patients - relevant when a child with stippled epiphyses also has neurological signs, and a reminder that this is a peroxisomal leukoencephalopathy rather than a purely skeletal condition. Diagnosis of any peroxisomal disorder rests on a battery of blood and urine assays confirmed in cultured fibroblasts and by DNA analysis, and treatment remains largely symptomatic.
Do not answer "rhizomelic CDP is lethal in infancy" flatly. Say it is a spectrum graded by residual plasmalogen: classic disease with negligible plasmalogens is severe, while nonclassic RCDP has higher plasmalogens, better growth and better cognition. Five genetic types are recognised - PEX7, GNPAT, AGPS, FAR1 and PEX5L - and no reliable survival figure for the group was retrievable.
Management
- Diagnosis: identify the form - genetic/metabolic testing (peroxisomal studies for rhizomelic CDP; EBP/ ARSL for X-linked forms) and a maternal drug/autoimmune history (warfarin, SLE).
- Orthopaedic: manage limb-length discrepancy (epiphysiodesis/lengthening), scoliosis and contractures in survivable forms (especially Conradi-Hunermann).
- Multidisciplinary: ophthalmology (cataracts), dermatology (ichthyosis), metabolic/genetics and family counselling.
- Severe rhizomelic form: largely supportive/palliative given the poor prognosis.
The radiographic stippling of the epiphyses identifies chondrodysplasia punctata as a group, but it does not by itself determine prognosis or management, because the forms differ enormously. The rhizomelic form is a severe autosomal-recessive peroxisomal disorder with symmetric proximal-limb shortening, developmental delay and cataracts, and in its classic form is often lethal in early childhood, so care is largely supportive - though nonclassic patients with higher residual plasmalogen levels do considerably better, which is why the biochemistry should be known before a family is counselled. Conradi-Hunermann (X-linked dominant, EBP) is milder and survivable, and is recognised by its asymmetry - asymmetric limb shortening with limb-length discrepancy, scoliosis, and ichthyosis following Blaschko's lines - which generates real orthopaedic work in deformity and discrepancy correction. Crucially, important secondary/teratogenic phenocopies exist: maternal warfarin in pregnancy (warfarin embryopathy) and maternal autoimmune disease such as systemic lupus erythematosus can produce stippled epiphyses, so a maternal drug and autoimmune history must be taken. Accurate classification - genetic/metabolic testing plus the maternal history - therefore guides both prognosis and the multidisciplinary management.
The Metabolic Basis of Each Form
- Rhizomelic CDP - a plasmalogen defect. A peroxisomal disorder of plasmalogen (ether-phospholipid) biosynthesis: RCDP1 = PEX7 (the PTS2 receptor importing the plasmalogen-synthesis enzymes into the peroxisome), RCDP2-4 = GNPAT, AGPS and FAR1 (the enzymes themselves), and RCDP5 = loss of the long isoform of PEX5 (PEX5L), which co-receptors PTS2 import and so mimics PEX7 loss. The result is deficient plasmalogens (with raised phytanic acid in type 1) - low red-cell plasmalogens and raised phytanic acid confirm it biochemically.
- Conradi-Hunermann - a cholesterol-synthesis defect. EBP encodes emopamil-binding protein, a sterol Δ8-Δ7 isomerase in the post-squalene cholesterol biosynthesis pathway; its deficiency causes accumulation of sterol precursors (8-dehydrocholesterol, 8(9)-cholestenol - measurable in plasma). The male lethality reflects the essential cholesterol pathway, and the Blaschko-line skin reflects X-inactivation mosaicism in surviving females.
- Why the epiphyses are stippled. The punctate calcification is abnormal/ectopic calcific deposition within the un-ossified epiphyseal cartilage, which resolves as the cartilage ossifies normally with growth.
Q: What metabolic pathways underlie the rhizomelic and Conradi-Hünermann forms, and why are the epiphyses stippled?
A: Rhizomelic CDP = a peroxisomal plasmalogen-synthesis defect across five types (RCDP1 PEX7 [the PTS2 receptor], RCDP2-4 GNPAT/AGPS/FAR1, RCDP5 PEX5L) → low plasmalogens + raised phytanic acid (biochemical diagnosis), with the residual plasmalogen level grading severity. Conradi-Hünermann = EBP (emopamil-binding protein), a sterol Δ8-Δ7 isomerase in post-squalene cholesterol synthesis → accumulated sterol precursors (8-dehydrocholesterol); male-lethal, Blaschko-line skin from X-inactivation mosaicism. The stippling = ectopic calcification in un-ossified epiphyseal cartilage that resolves as it ossifies.
Mnemonics & Memory Aids
STIPPLE
Hook:STIPPLE: Stippled epiphyses, Teratogenic phenocopies, Ichthyosis/cataracts, Peroxisomal rhizomelic (severe), Pattern (symmetric vs asymmetric), Limb sequelae, EBP gene.
Clinical Decision Scenarios
Practise clinical reasoning and management decisions out loud
“An infant radiograph shows stippled epiphyses. What is the diagnosis group, what forms do you consider, and what history is important?”
Hallmark
- Stippled (punctate) epiphyseal/peri-articular calcification in infancy
- Heterogeneous group; stippling resolves with growth
- Identify the form (prognosis/management differ greatly)
Genetic forms
- Rhizomelic CDP: AR peroxisomal plasmalogen defect, RCDP1-5 (PEX7, GNPAT, AGPS, FAR1, PEX5L)
- A SPECTRUM: classic (negligible plasmalogens) severe with early death; nonclassic (higher) milder - the level is prognostic
- Conradi-Hunermann (CDPX2): X-linked dominant (EBP), females, asymmetric shortening + ichthyosis + cataracts
- X-linked recessive (CDPX1, ARSL): brachytelephalangic (nasal/distal-phalangeal hypoplasia)
Secondary/teratogenic
- Warfarin embryopathy (maternal warfarin in pregnancy)
- Maternal autoimmune disease (e.g. SLE)
- Take a maternal drug/autoimmune history
Orthopaedic management
- Limb shortening/discrepancy (epiphysiodesis/lengthening)
- Scoliosis and contractures (survivable forms)
- Multidisciplinary: ophthalmology (cataracts), dermatology, genetics; severe form palliative
Evidence & Key Studies
Skeletal dysplasias of the fetus and infant (including chondrodysplasia punctata)
- More than 400 genetic skeletal disorders are included in the latest classification; the most severe/lethal phenotypes are identifiable in the prenatal period, and perinatal autopsy/post-mortem radiographs are crucial for definitive diagnosis.
- Chondrodysplasia punctata is among the recognised skeletal dysplasias encountered in fetal/neonatal practice (alongside thanatophoric dysplasia, osteogenesis imperfecta, campomelic dysplasia and achondroplasia).
- An increasing number of cases are confirmed by genetic testing, reflecting the heterogeneous genetic basis of these dysplasias.
Plasmalogen level grades the rhizomelic phenotype - classic against nonclassic disease
- RCDP type 1 follows PEX7 defects impairing plasmalogen biosynthesis and phytanic acid oxidation; plasmalogen deficiency is described as the main pathogenic factor determining the SEVERITY of the disease.
- Severe (classic) patients have negligible plasmalogen levels, congenital cataracts, skeletal dysplasia, growth and neurodevelopmental deficits, with cerebral hypomyelination and cerebellar atrophy on MRI.
- Milder or NONCLASSIC RCDP patients have higher plasmalogen levels, better growth and better cognitive outcomes - so the condition is a graded spectrum rather than a uniformly lethal disease.
- An allelic mouse series reproduced the genotype-phenotype gradient, with incremental increases in Pex7 producing marked phenotypic improvement.
- The experimental work is in mice; the classic-versus-nonclassic human distinction is drawn from the clinical literature the authors summarise rather than from a human cohort reported here.
RCDP5: losing only the long isoform of PEX5 causes rhizomelic disease, not Zellweger spectrum
- Four patients from two independent families with a homozygous frameshift in PEX5 exon 9, which is specific to the long isoform PEX5L.
- Other PEX5 mutations cause Zellweger spectrum disorders through failed PTS1 import; losing PEX5L alone impairs import of PTS2-tagged proteins only - the same functional defect as PEX7 loss - and therefore produces RCDP.
- Establishes RCDP5, alongside RCDP1 (PEX7) and RCDP2 to 4 (GNPAT, AGPS, FAR1).
- Because RCDP1 and RCDP5 overlap biochemically, both PEX7 and PEX5 exon 9 should be sequenced when there is a selective PTS2-import defect.
- Four patients: it defines a genetic type and says nothing about how common it is.
The recognition of chondrodysplasia punctata among the genetic skeletal dysplasias of the fetus/infant, the breadth of the dysplasia classification, and the role of radiographs/genetic testing in diagnosis come from the cited Jezova review. The unifying radiographic hallmark (stippled epiphyses resolving with age), the specific forms (rhizomelic peroxisomal; Conradi-Hunermann X-linked dominant EBP with asymmetry, ichthyosis and cataracts; X-linked recessive brachytelephalangic), the teratogenic phenocopies (warfarin embryopathy, maternal autoimmune disease), and the orthopaedic sequelae/management are standard, well-established teaching. The classic-versus-nonclassic gradation of rhizomelic disease by residual plasmalogen level, and the brain MRI findings, come from the cited Fallatah paper, whose experimental work is in mice and whose human distinction summarises the clinical literature rather than reporting a cohort. The existence of RCDP5 and the advice to sequence PEX5 exon 9 alongside PEX7 come from the cited Baroy report of four patients from two families, which cannot indicate how common that type is. No reliable survival figure, age-at-death distribution or proportion falling into the nonclassic group was retrieved for rhizomelic CDP, so none is quoted - the prognosis is described qualitatively and tied to the biochemistry instead. See also skeletal dysplasias, limb-length discrepancy and epiphysiodesis and congenital scoliosis; the library has no general scoliosis topic under that name.